Navigation Menu
Search code, repositories, users, issues, pull requests..., provide feedback.
We read every piece of feedback, and take your input very seriously.

Saved searches
Use saved searches to filter your results more quickly.
To see all available qualifiers, see our documentation .
- Notifications You must be signed in to change notification settings
Applications of machine learning in perovskite materials
- Published: 30 September 2022
- Volume 5 , pages 2700–2720, ( 2022 )
Cite this article

- Ziman Wang 1 , 6 na1 ,
- Ming Yang 1 , 6 na1 ,
- Xixi Xie 1 , 6 ,
- Chenyang Yu 1 , 6 ,
- Qinglong Jiang 2 ,
- Mina Huang 3 , 4 ,
- Hassan Algadi 5 ,
- Zhanhu Guo 4 &
- Hang Zhang 1 , 6
3579 Accesses
44 Citations
Explore all metrics
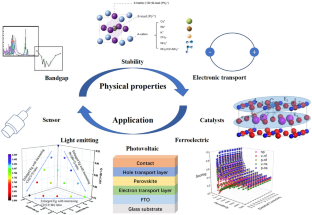
Machine learning (ML) offers the opportunities to discover certain unique properties for typical material. Taking perovskite materials as an example, this review summarizes the applications of ML in predicting their bandgap, stability, electronic transport, catalytic, ferroelectric, photovoltaic, light emitting, and sensing properties. This proves that ML can accelerate the screening and discovery of novel materials with potential properties for certain applications.
This is a preview of subscription content, log in via an institution to check access.
Access this article
Price includes VAT (Russian Federation)
Instant access to the full article PDF.
Rent this article via DeepDyve
Institutional subscriptions
Similar content being viewed by others

Advances in the Application of Perovskite Materials

Two-Dimensional Materials for Highly Efficient and Stable Perovskite Solar Cells

Predictive machine learning approaches for perovskites properties using their chemical formula: towards the discovery of stable solar cells materials
Kojima A, Teshima K, Shirai Y et al (2009) Organometal halide perovskites as visible-light sensitizers for photovoltaic cells. J Am Chem Soc 131:6050–6051. https://doi.org/10.1021/ja809598r
Article CAS Google Scholar
Green MA, Dunlop ED, Hohl-Ebinger J et al (2019) Solar cell efficiency tables (Version 55). Prog Photovolt 28:3–15. https://doi.org/10.1002/pip.3228
Article Google Scholar
Rostalski J, Meissner D (2000) Monochromatic versus solar efficiencies of organic solar cells. Sol Energy Mater Sol Cells 61:87–95. https://doi.org/10.1016/S0927-0248(99)00099-9
Mickelsen RA, Chen S (1981) Development of a 9.4% efficient thin-film CuInSe 2 /CdS solar cell. Proc 15th IEEE Photovolt Special Conf 15:800–804
Kamada R, Yagioka T, Adachi S et al (2016) New world record Cu(In,Ga)(Se,S) 2 Thin film solar cell efficiency beyond 22%. Proc 43rd IEEE Photovoltaic Specialists Conference 1287–1291
Blakers A, Zin N, McIntosh KR et al (2013) High efficiency silicon solar cells. PV Asia Pacific Conference 33:1–10. https://doi.org/10.1016/j.egypro.2013.05.033
Liu K, Yang H, Wang W et al (2021) Numerical and experimental exploration towards a 26% efficiency rear-junction n-type silicon solar cell with front local-area and rear full-area polysilicon passivated contacts. Sol Energy 221:1–9. https://doi.org/10.1016/j.solener.2021.04.020
Tao Q, Xu P, Li M et al (2021) Machine learning for perovskite materials design and discovery. NPJ Comput Mater 7. https://doi.org/10.1038/s41524-021-00495-8
Tanaka I (2020) Data-driven materials discovery from large chemistry spaces. Matter 3:327–328. https://doi.org/10.1016/j.matt.2020.07.010
Zhang H, Hippalgaonkar K, Buonassisi T et al (2018) Machine learning for novel thermal-materials discovery: early successes, opportunities, and challenges. ES Energy & Environment 2:1–8. https://doi.org/10.30919/esee8c209
Kumbhar A, Dhawale PG, Kumbhar S et al (2021) A comprehensive review: machine learning and its application in integrated power system. Energ Rep 7:5467–5474. https://doi.org/10.1016/j.egyr.2021.08.133
Zhou ZH (2016) Machine Learning. Tsinghua University Press, Beijing
Google Scholar
Li C, Hao H, Xu B et al (2020) A progressive learning method for predicting the band gap of ABO 3 perovskites using an instrumental variables. J Mater Chem C 8:3127–3136. https://doi.org/10.1039/c9tc06632b
Allam O, Holmes C, Greenberg Z et al (2018) Density functional theory-machine learning approach to analyze band gap of elemental halide perovskites and Ruddlesden-Popper Phases. Chemphyschem 19:2559–2565. https://doi.org/10.1002/cphc.201800382
Park H, Mall R, Ali A et al (2020) Importance of structural deformation features in the prediction of hybrid perovskite bandgaps. Comput Mater Sci 184:109858. https://doi.org/10.1016/j.commatsci.2020.109858
Pilania G, Mannodi-Kanakkithodi A, Uberuaga BP et al (2016) Machine learning bandgaps of double perovskites. Sci Rep 6. https://doi.org/10.1038/srep19375
Yang X, Li L, Tao Q et al (2021) Rapid discovery of narrow bandgap oxide double perovskites using machine learning. Comput Mater Sci 196:110528. https://doi.org/10.1016/j.commatsci.2021.110528
Chaube S, Khullar P, Srinvasan SG et al (2019) A statistical learning framework for accelerated bandgap prediction of inorganic compounds. J Electron Mater 49:752–762. https://doi.org/10.1007/s11664-019-07779-2
Omprakash P, Manikandan B, Sandeep A et al (2021) Graph representational learning for bandgap prediction in varied perovskite crystals. Comput Mater Sci 196:110530. https://doi.org/10.1016/j.commatsci.2021.110530
Li W, Jacobs R, Morgan D (2018) Predicting the thermodynamic stability of perovskite oxides using machine learning models. Comput Mater Sci 150:454–463. https://doi.org/10.1016/j.commatsci.2018.04.033
Liu H, Cheng J, Dong H et al (2020) Screening stable and metastable ABO 3 perovskites using machine learning and the materials project. Comput Mater Sci 177:109614. https://doi.org/10.1016/j.commatsci.2020.109614
Talapatra A, Uberuaga BP, Stanek CR et al (2021) A machine learning approach for the prediction of formability and thermodynamic stability of single and double perovskite oxides. Chem Mater 33:845–858. https://doi.org/10.1021/acs.chemmater.0c03402
Maddah HA, Berry V, Behura SK (2020) Cuboctahedral stability in titanium halide perovskites via machine learning. Comput Mater Sci 173:109415. https://doi.org/10.1016/j.commatsci.2019.109415
Zhao Y, Zhang J, Xu Z et al (2021) Discovery of temperature-induced stability reversal in perovskites using high-throughput robotic learning. Nat Commun 12:2191. https://doi.org/10.1038/s41467-021-22472-x
Hillhouse HW, Dunlap-Shohl WA, Stoddard RJ (2020) Quantitative prediction of perovskite stability using accelerated testing and machine learning. 47th IEEE Photovoltaic Specialists Conference:2116–2119. https://doi.org/10.1109/PVSC45281.2020.9300931
Roekeghem A, van Carrete J, Oses C et al (2016) High-throughput computation of thermal conductivity of high-temperature solid phases: the case of oxide and fluoride perovskites. Phys Rev X 6. https://doi.org/10.1103/PhysRevX.6.041061
Schmidt J, Shi J, Borlido P et al (2017) Predicting the thermodynamic stability of solids combining density functional theory and machine learning. Chem Mater 29:5090–5103. https://doi.org/10.1021/acs.chemmater.7b00156
Yang M, Zhang X, Zhang H (2021) Effects of monovacancy on thermal properties of bilayer graphene nanoribbons by molecular dynamics simulations. J Therm Sci:1–8. https://doi.org/10.1007/s11630-021-1412-9
Wang Z, Yang M, Jiang Q et al (2022) Improving the thermoelectric properties of 2,7-dioctyl[1]benzothieno[3,2-b][1]benzothiophene-based organic semiconductors by isotropic strain. ES Mater Manuf 16:66–77. https://doi.org/10.30919/esmm5f489
Priya P, Aluru NR (2021) Accelerated design and discovery of perovskites with high conductivity for energy applications through machine learning. NPJ Comput Mater 7. https://doi.org/10.1038/s41524-021-00551-3
Liu X, Lu W, Peng C et al (2009) Two semi-empirical approaches for the prediction of oxide ionic conductivities in ABO 3 perovskites. Comput Mater Sci 46:860–868. https://doi.org/10.1016/j.commatsci.2009.04.047
Li L, You Y, Hu S et al (2019) Electronic transport of organic-inorganic hybrid perovskites from first-principles and machine learning. Appl Phys Lett 114:083102. https://doi.org/10.1063/1.5045512
Zhou G, Chu W, Prezhdo OV (2020) Structure deformation controls charge losses in MAPbI 3 : unsupervised machine learning of nonadiabatic molecular dynamics. ACS Energy Lett 5:1930–1938. https://doi.org/10.1021/acsenergylett.0c00899
Li Z, Achenie LEK, Xin H (2020) An adaptive machine learning strategy for accelerating discovery of perovskite electrocatalysts. ACS Catal 10:4377–4384. https://doi.org/10.1021/acscatal.9b05248
Wang X, Xiao B, Li Y et al (2020) First-principles based machine learning study of oxygen evolution reactions of perovskite oxides using a surface center-environment feature model. Appl Surf Sci 531:147323. https://doi.org/10.1016/j.apsusc.2020.147323
Tao Q, Lu T, Sheng Y (2021) Machine learning aided design of perovskite oxide materials for photocatalytic water splitting. J Energ Chem 60:351–359. https://doi.org/10.1016/j.jechem.2021.01.035
Weng B, Song Z, Zhu R et al (2020) Simple descriptor derived from symbolic regression accelerating the discovery of new perovskite catalysts. Nat Commun: 11. https://doi.org/10.1038/s41467-020-17263-9
Zhang G, Fan B, Zhao P et al (2018) Ferroelectric polymer nanocomposites with complementary nanostructured fillers for electrocaloric cooling with high power density and great efficiency. ACS Appl Energy Mater (3):1344–1354. https://doi.org/10.1021/acsaem.8b00052
Wang Z, Gao Y, Ma Y et al (2021) Enhanced electrocaloric effect within a broad temperature range in lead-free polymer composite films by blending the rare-earth doped BaTiO 3 nanopowders. Adv Comp Hybr Mater 4(3):469–477. https://doi.org/10.1007/s42114-021-00252-x
Wang Z, Yang M, Zhang H (2021) Strain engineering on electrocaloric effect in PbTiO 3 and BaTiO 3 . Adv Comp Hybr Mater 4(4):1239–1247. https://doi.org/10.1007/s42114-021-00257-6
Min K, Cho E (2020) Accelerated discovery of potential ferroelectric perovskite via active learning. J Mater Chem C 8(23). https://doi.org/10.1039/d0tc00985g
Ziatdinov M, Nelson CT, Zhang X et al (2020) Causal analysis of competing atomistic mechanisms in ferroelectric materials from high-resolution scanning transmission electron microscopy data. NPJ Comput Mater 6(1):127. https://doi.org/10.1038/s41524-020-00396-2
He J, Li J, Liu C et al (2021) Machine learning identified materials descriptors for ferroelectricity. Acta Mater 209:116815. https://doi.org/10.1016/j.actamat.2021.116815
Kim C, Pilania G, Ramprasad R (2016) Machine learning assisted predictions of intrinsic dielectric breakdown strength of ABX 3 perovskites. J Phys Chem C 120:14575–14580. https://doi.org/10.1021/acs.jpcc.6b05068
Shen Z, Bao Z, Cheng X et al (2021) Designing polymer nanocomposites with high energy density using machine learning. NPJ Comput Mater 7:110. https://doi.org/10.1038/s41524-021-00578-6
Zhai X, Chen M, Lu W (2018) Accelerated search for perovskite materials with higher Curie temperature based on the machine learning methods. Comput Mater Sci 151:41–48. https://doi.org/10.1016/j.commatsci.2018.04.031
Balachandran PV, Kowalski B, Sehirlioglu A et al (2018) Experimental search for high-temperature ferroelectric perovskites guided by two-step machine learning. Nat Commun 9(1):1668. https://doi.org/10.1038/s41467-018-03821-9
Yilmaz B, Yildirim R (2021) Critical review of machine learning applications in perovskite solar research. Nano Energy 80:105546. https://doi.org/10.1016/j.nanoen.2020.105546
Geng W, Tong C, Zhang Y et al (2020) Theoretical progress on the relationship between the structures and properties of perovskite solar cells. Adv Theory Simul. https://doi.org/10.1002/adts.202000022
He Z, Yang M, Wang L et al Concentrated photovoltaic thermoelectric hybrid system: an experimental and machine learning study. Engi Sci 15:47–56. https://doi.org/10.30919/es8d440
Chen C, Xie X, Yang M et al (2021) ES Energy Environ 11:3–18. https://doi.org/10.30919/esee8c416
Jin H, Zhang H, Li J et al (2020) Discovery of Novel Two-Dimensional Photovoltaic Materials Accelerated by Machine Learning. J Phys Chem Lett 11:3075–3081. https://doi.org/10.1021/acs.jpclett.0c00721
Lu S, Zhou Q, Ma L et al (2019) Rapid discovery of ferroelectric photovoltaic perovskites and material descriptors via machine learning. Small Methods 3(11). https://doi.org/10.1002/smtd.201900360
Srivastava M, Howard J M, Gong T et al (2021) Machine learning roadmap for perovskite photovoltaics. J Phys Chem Lett 12:7866–7877. https://doi.org/10.1021/acs.jpclett.1c01961
Nelson MD, Vece MD (2019) Using a neural network to improve the optical absorption in halide perovskite layers containing core-shells silver nanoparticles. Nanomater 9(3). https://doi.org/10.3390/nano9030437
Agiorgousis ML, Sun Y, Choe DH et al (2019) Machine learning augmented discovery of chalcogenide double perovskites for photovoltaics. Adv Theory Simul 2:1800173. https://doi.org/10.1002/adts.201800173
Hartono NTP, Thapa J, Tiihonen A et al (2020) capping layers design guidelines for stable perovskite solar cells via machine learning. 47th IEEE Photovoltaic Specialists Conference:693–695. https://doi.org/10.1109/PVSC45281.2020.9300622
Li J, Pradhan B, Gaur S et al (2019) Predictions and strategies learned from machine learning to develop high-performing perovskite solar cells. Adv Energy Mater 9(46). https://doi.org/10.1002/aenm.201901891
Takahashi K, Takahashi L, Miyazato I et al (2018) Searching for hidden perovskite materials for photovoltaic systems by combining data science and first principle calculations. ACS Photonics 5:771–775. https://doi.org/10.1021/acsphotonics.7b01479
Gladkikh V, Kim DY, Hajibabaei A et al (2020) Machine learning the band gaps of ABX 3 Perovskites from Elemental Properties. J Phys Chem C 124:8905–8918. https://doi.org/10.1021/acs.jpcc.9b11768
Li Y, Lu Y, Huo X et al (2021) Bandgap tuning strategy by cations and halide ions of lead halide perovskites learned from machine learning. RSC Adv 11:15688–15694. https://doi.org/10.1039/d1ra03117a
Heimbrook A, Higgins K, Kalinin SV et al (2021) Exploring the physics of cesium lead halide perovskite quantum dots via Bayesian inference of the photoluminescence spectra in automated experiment. Nanophotonics 10:1977–1989. https://doi.org/10.1515/nanoph-2020-0662 .
Li Z, Xu Q, Sun Q et al (2019) Thermodynamic Stability Landscape of Halide Double Perovskites via High-Throughput Computing and Machine Learning. Adv Funct Mater 29:1807280. https://doi.org/10.1002/adfm.201807280
Lu S, Zhou Q, Ouyang Y et al (2018) Accelerated discovery of stable lead-free hybrid organic-inorganic perovskites via machine learning. Nat Commun 9:3405. https://doi.org/10.1038/s41467-018-05761-w
Shao S, Xie C, Zhang L et al (2021) CsPbI 3 NC-sensitized SnO 2 /multiple-walled carbon nanotube self assembled nanomaterials with highly selective and sensitive NH 3 sensing performance at room temperature. ACS Appl Mater Interfaces 13:14447–14457. https://doi.org/10.1021/acsami.0c20566
Liu T, He J, Lu Z et al (2022) A visual electrochemiluminescence molecularly imprinted sensor with Ag + @UiO-66-NH 2 decorated CsPbBr 3 perovskite based on smartphone for point-of-care detection of nitrofurazone. Chem Eng J 429:132462. https://doi.org/10.1016/j.cej.2021.132462
Download references
This work is supported by the Basic Science Center Program for Ordered Energy Conversion of the National Natural Science Foundation of China (No.51888103, 51606192) and NIH-Arkansas INBRE.
Author information
Ziman Wang and Ming Yang contribute equally to the article.
Authors and Affiliations
Institute of Engineering Thermophysics, Chinese Academy of Sciences, Beijing, 100190, China
Ziman Wang, Ming Yang, Xixi Xie, Chenyang Yu & Hang Zhang
Department of Chemistry and Physics, University of Arkansas, Pine Bluff, AR, 71601, USA
Qinglong Jiang
College of Materials Science and Engineering, Taiyuan University of Science and Technology, Taiyuan, 030024, China
Integrated Composites Laboratory (ICL), Department of Chemical and Bimolecular Engineering, University of Tennessee, Knoxville, TN, 37996, USA
Mina Huang & Zhanhu Guo
Department of Electrical Engineering, Faculty of Engineering, Najran University, P.O. Box 1988, Najran, 11001, Saudi Arabia
Hassan Algadi
University of Chinese Academy of Sciences, Beijing, 100049, China
You can also search for this author in PubMed Google Scholar
Contributions
Zhanhu Guo and Hang Zhang conceived and designed the review. Ziman Wang, Ming Yang, Xixi Xie, Chenyang Yu, and Qinglong Jiang wrote the main text. Mina Huang and Hassan Algadi prepared figures. All authors read and approved the manuscript.
Corresponding authors
Correspondence to Qinglong Jiang , Zhanhu Guo or Hang Zhang .
Ethics declarations
Conflict of interest.
The authors declare no competing interests.
Additional information
Publisher's note.
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
Reprints and permissions
About this article
Wang, Z., Yang, M., Xie, X. et al. Applications of machine learning in perovskite materials. Adv Compos Hybrid Mater 5 , 2700–2720 (2022). https://doi.org/10.1007/s42114-022-00560-w
Download citation
Received : 13 June 2022
Revised : 02 September 2022
Accepted : 09 September 2022
Published : 30 September 2022
Issue Date : December 2022
DOI : https://doi.org/10.1007/s42114-022-00560-w
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
- Machine learning
- Perovskite materials
- Physical properties
- Application research
- Find a journal
- Publish with us
- Track your research
Thank you for visiting nature.com. You are using a browser version with limited support for CSS. To obtain the best experience, we recommend you use a more up to date browser (or turn off compatibility mode in Internet Explorer). In the meantime, to ensure continued support, we are displaying the site without styles and JavaScript.
- View all journals
- My Account Login
- Explore content
- About the journal
- Publish with us
- Sign up for alerts
- Open access
- Published: 26 February 2019
Bandgap prediction by deep learning in configurationally hybridized graphene and boron nitride
- Yuan Dong 1 na1 ,
- Chuhan Wu 2 na1 ,
- Chi Zhang 1 ,
- Yingda Liu 2 ,
- Jianlin Cheng 2 &
- Jian Lin ORCID: orcid.org/0000-0002-4675-2529 1
npj Computational Materials volume 5 , Article number: 26 ( 2019 ) Cite this article
12k Accesses
89 Citations
71 Altmetric
Metrics details
- Atomistic models
- Electrical and electronic engineering
- Electronic properties and devices
- Two-dimensional materials
It is well-known that the atomic-scale and nano-scale configuration of dopants can play a crucial role in determining the electronic properties of materials. However, predicting such effects is challenging due to the large range of atomic configurations that are possible. Here, we present a case study of how deep learning algorithms can enable bandgap prediction in hybridized boron–nitrogen graphene with arbitrary supercell configurations. A material descriptor that enables correlation of structure and bandgap was developed for convolutional neural networks. Bandgaps calculated by ab initio calculations, and corresponding structures, were used as training datasets. The trained networks were then used to predict bandgaps of systems with various configurations. For 4 × 4 and 5 × 5 supercells they accurately predict bandgaps, with a R 2 of >90% and root-mean-square error of ~0.1 eV. The transfer learning was performed by leveraging data generated from small supercells to improve the prediction accuracy for 6 × 6 supercells. This work will pave a route to future investigation of configurationally hybridized graphene and other 2D materials. Moreover, given the ubiquitous existence of configurations in materials, this work may stimulate interest in applying deep learning algorithms for the configurational design of materials across different length scales.
Similar content being viewed by others

A quantum coherent spin in hexagonal boron nitride at ambient conditions

Reproducible graphene synthesis by oxygen-free chemical vapour deposition

Non-trivial quantum geometry and the strength of electron–phonon coupling
Introduction.
Introduction of defects at atomic or nanoscales has been a widely employed strategy in bulk materials, such as metals, ceramics, and semiconductors, for manipulation of their mechanical and physical properties. Especially when these defects are precisely controlled in configuration space, they significantly improve the mechanical properties, 1 , 2 , 3 manipulate the magnetic properties, 4 , 5 , 6 and alter the electronic properties. 7 , 8 , 9 , 10 For instance, nanotwined grain boundaries are crucial to realize ultrahigh strength, superior fatigue resistance in metals, 1 and ultrahigh hardness and enhanced toughness in ceramics. 2 , 3 Localization of nitrogen vacancy center at deterministic locations with nanoscale precision within diamond improves sensitivity and resolution of single electron spin imaging, 4 , 5 and enhances charge-state switching rates, 6 paving new ways to next-generation quantum devices. Precisely controlling dopant atoms in term of concentration and spatial arrangement within semiconductors is so critical to performance of fabricated electronic devices, especially as the dimension of the device keeps shrinking. 7 Manipulation and detection of individual surface vacancies in a chlorine terminated Cu (100) surface realizes atomic scale memory devices. 10
Based on the aforementioned examples in the bulk materials, it is naturally anticipated that the effect of the defects in two-dimensional (2D) materials, in term of the defect density and configurations, would be even more profound as all the atoms are confined within a basal plane with atomic thickness. Distinguished from allowance of different pathways of defect configurations in the three-dimensional (3D) bulk materials, this dimensionality restriction in the 2D materials largely reduces the accessibility and variability of the defects. This uniqueness would allow configurational design of the defects in 2D, which starts to emerge as a new and promising research field. As the first well-known 2D material, graphene has been shown to exhibit configurational grain boundaries-dependent mechanical, 11 , 12 thermal, 13 and electrical properties. 14 , 15 Doping graphene with heteroatoms, such as hydrogen, nitrogen, and boron, can further tailor the magnetic or electrical properties. 16 Theoretical calculations suggest that these properties not only depend on types of dopants, 16 , 17 , 18 and doping concentrations, 19 but also are greatly determined by the dopants configurations within the graphene. 20 , 21 , 22 Although the theoretical investigation already enables to research a much larger set of cases than the experiment does. The number of possible configurations for the dopants in graphene far exceeds the amount that can be practically computed due to extremely high computational cost. For instance, hybridizing boron–nitrogen (B–N) pairs into a graphene layer with a just 6 × 6 supercell system results in billions of possible configuration structures. Thus, it is entirely impractical to study all the possibilities to get the optimized properties even for such a small system. Another limitation of current mainstream of material design is that it heavily relies on intuition and knowledge of human who design, implement, and characterize materials through the trial and error processes.
Recent progress in data-driven machine learning (ML) starts to stimulate great interests in material fields. For instance, a series of material properties of stoichiometric inorganic crystalline materials were predicted by the ML. 23 In addition to their potentials in predicting properties of the materials, they start to show great power in assisting materials design and synthesis. 24 , 25 , 26 , 27 It is anticipated that ML would assist to push the material revolution to a paradigm of full autonomy in the next 5–10 years, 28 , 29 especially as emerging of deep learning (DL) algorithms. 30 , 31 For instance, a pioneer work of employing only a few layered convolutional neural networks (CNNs) enables to reproduce the phase transition of matters. 32 Nevertheless, the application of the DL in the material fields is still in its infancy. 33 One of main barriers is that a compatible and sophisticated descriptive system that enables to correlate the predicted properties to structures is required for materials because DL algorithms are originally developed for imaging recognition.
Motivated by this challenge, we conceive to employ CNNs, including VGG16 convolutional network (VCN), residual convolutional network (RCN), and our newly-constructed concatenate convolutional network (CCN), for predicting electronic properties of hybridized graphene and h-BN with randomly configured supercells. As a benchmark comparison, the support vector machine (SVM), 34 which used to be the mainstream ML algorithm before the DL era, was also adopted (see details in Supplementary Note 1 ). We discovered that after trained with structural information and the bandgaps calculated from ab initio density function theory (DFT), these CNNs enabled to precisely predict the bandgaps of hybridized graphene and boron nitride (BN) pairs with any given configurations. One main reason for the high prediction accuracy arises from the developed material descriptor. Such a descriptive system enables to qualitatively and quantitatively capture the features of configurational states, where each atom in the structure affects its neighbor atoms so that these localized atomic clusters collectively determine bandgaps of the whole structure. Combined with well-tuned hyperparameters and well-designed descriptor, the CNNs result in high prediction accuracy. Considering that atom-scale precise structures of doped graphene by bottom-up chemical synthesis have been experimental realized, 35 , 36 , 37 this work provides a cornerstone for future investigation of graphene and other 2D materials as well as their associated properties. We believe that this work will bring up broader interests in applying the designed descriptive system and the CNN models for many materials related problems, which are not accessible to other ML algorithms.
Results and discussion
Dataset for bandgaps and structures of configurationally hybridized graphene.
The generation of various configurations follows a pure geometric way. Because graphene and h-BN have similar honeycomb structures with very close bond lengths (1.42 Å and 1.44 Å for graphene and h-BN, respectively), 38 which is beneficial to structural stability if B–N pairs are hybridized in graphene. Moreover, graphene is a semimetal with a zero bandgap while h-BN is a wide-bandgap semiconductor. Thus, it can be naturally assumed that the graphene hybridized with the B–N pairs could have an intermediate bandgap. Finally, the B–N pairs can exactly make the charge neutral in the doped graphene. To test these hypotheses, we first implemented high throughput DFT calculations on configurationally hybridized graphene to generate datasets—correlation of supercell structures to bandgaps—for the ML. To achieve high-throughput calculations, we only apply non-hybrid function for the DFT calculations. Even though the calculated bandgaps are not as accurate as the previously reported values calculated by the hybrid functions, data consistence can be secured. Initial calculations show that the bandgaps of the pristine graphene with 4 × 4, 5 × 5, and 6 × 6 supercell systems are exactly 0 eV (Fig. 1a ), and the bandgap of the pristine h-BN is 4.59 eV (Fig. 1b ). More DFT calculations on examples of 4 × 4 systems that have the 50 at% B–N concentrations but with different configurational states show that they exhibit bandgaps ranging from 0.95 to 1.57 eV (Fig. 1c ). As the BN concentration increases to 62.5 at%, the corresponding bandgaps also increase (Fig. 1d ). The band structure of 6 × 6 supercell systems of graphene doped with 3 at% B–N concentrations (Fig. S1 ) illustrates the dopant induced bands/levels. These results validate that bandgaps of hybridized graphene depend on the configurations. Then more bandgaps of hybrids with arbitrary concentration and supercell configurations were calculated by the DFT. Some of them served as the training datasets for training the CNNs. Others served as test datasets to validate the accuracy of the prediction performed by the CNNs.

a , b Band structures of pristine graphene a and pristine h-BN b . c , d Representative atomic configurations of 4 × 4 graphene supercell systems that have the same B-N dopant concentrations (50% and 62.5%) but with different configurational states and their corresponding bandgaps. C, N, B atoms are colored with black, blue, and red
After the bandgap matrix is generated, structural information, such as chemical compositions and structures, needs to be well described in a form of numerical matrixes which will serve input data to train the CNNs. Defining descriptors of the materials for the ML is one of main challenges because the descriptors are more important to influence model accuracy than the ML algorithms do. 23 , 39 Herein, we chose a simple and illustrative descriptor which is very suitable for the DL framework. We define the hybridized graphene structure by a 2D matrix described with only “0” and “1”. “0” corresponds to a C–C pair while “1” corresponds to a B–N pair (Fig. 2a ). The size of matrix is 4 × 4, 5 × 5, and 6 × 6, the same as the size of supercells. This natural representation like a 2D image significantly simplifies the learning process, securing the model accuracy. Note that we exclude the cases of switching configurations of B–N pairs, which will add more complexity to the investigated systems. The size of a matrix is determined by the size of a supercell in an investigated system. For instance, a 4 × 4 system was represented by a 4 × 4 matrix. In this work, 4 × 4, 5 × 5, and 6 × 6 systems were studied. To enlarge the training dataset, the equivalent structures were obtained by translating the particular structures along their lattice axis or inversion around their symmetry axis (Fig. S2 ). These structures are equivalent, thus having the same bandgaps. By this way, ~14,000, ~49,000, and ~7200 data examples for the 4 × 4, 5 × 5, and 6 × 6 supercell systems, respectively, were generated for purposes of training and validating CNNs. These datasets cover 21.36%, 0.15%, and 1 × 10 –7 of all possible configurations for the 4 × 4, 5 × 5, and 6 × 6 supercell systems, respectively. These datasets were split into the training and test datasets, respectively. For example, when investigating 4 × 4 supercell systems, we randomly chose 13,000 and 1000 different data points from all the data samples as the training and validation datasets, respectively, to train and validate the CNN models. Correspondingly, for 5 × 5 supercell systems, the training and validation datasets are 48,000 and 1000, respectively. For 6 × 6 supercell systems, the training and validation datasets are 6200 and 1000, respectively.

a Descriptors for 2D doped graphene supercell systems (4 × 4, 5 × 5, and 6 × 6 systems). b A convolutional neural network, VCN, for the prediction of bandgaps of 2D doped graphene systems
Construction of CNNs
The general procedure of setting up the CNNs is illustrated as follows. The structural information of the hybridized graphene/BN sheets is represented by input matrices. The input matrices are transformed into multiple feature maps by filters in the first convolutional layer. The output of the convolutional layer is further transformed into high-level feature maps by the next convolutional layer. The maximum value of each feature map of the last convolutional layer is pooled together by the max pooling, which is used as the input of the next three fully-connected layers (FC). The single node in the output layer takes the output of the last FC layer as input to predict the bandgaps. The CNNs used in this work were constructed into three different structures. Firstly, we constructed a network which is similar to the traditional VCN designed for image processing, 40 as shown in Fig. 2b . This network has 12 convolutional (Conv) layers, one global-flatten layer, three FC layers, and an output layer. The neural layers in VCN are explained in Supplementary Note 2 . The detailed hyperparameters are given in Table S1 .
Although the VCN is capable of learning from the data, our prediction results show that its performance is limited by the depth of neurons. The accuracy gets saturated and degraded rapidly. To tackle this problem, we further constructed the other two neural networks, RCN and CCN. The construction of RCN is explained in Supplementary Note 3 . Its structure and hyperparameters are shown in Fig. S3 and Table S2 – S4 . The structure of RCN that we used is similar to that of ResNet50 network, but the Max-Pooling layer was replaced with a Global Max pooling layer. The ResNet is a recent popular option in image classification. 41 A characteristic of this network is that the convolution layers in VCN are replaced with residual blocks. This design can prevent the degeneration when the network goes deeper. The block in the CCN is our newly-constructed network which combines advantages of both GoogleNet 42 and DenseNet. 43 The structure and hyperparameters of this network is explained in Supplementary Note 4 and illustrated in Fig. S4a and Table S5 - S6 . Similar to RCN, we also introduce the concatenation blocks into CCN as the building blocks (Fig. S4b and Table S6 ). Unlike RCN which “adds” feature maps in element-wise, CCN concatenates the layers from input and output data by simultaneously passing them through the activation layer. The concatenation of the network can also prevent it from degradation which is usually existing in the VCN. The concatenation can keep more extracted features in previous neuron layers, which would be beneficial to the transfer learning (TL). After the networks were built, they were trained by the generated datasets which correlate the structural information of the hybridized graphene with their corresponding bandgaps. After the CNNs were trained, 300 new datasets that illustrate 300 types of the hybridized graphene structures for each type of investigated supercell systems (4 × 4, 5 × 5, and 6 × 6) were fed into the trained CNNs to predict the bandgaps. Note that these used structures do not exist in ones used for training and validation. The predicted bandgaps were compared to the values calculated by the DFT on the same structures for evaluating the prediction accuracy of the DL algorithms.
Prediction of bandgaps by CNNs
As shown in Fig. 1 , we can conclude that the bandgaps of the hybridized graphene are influenced by both the dopants and their configurations. In other words, each atom in the structure affects its neighbor atoms so that these localized atomic clusters collectively determine bandgaps of the whole structure. As convolutions in the CNNs can extract the features not only from the elements of the input data but also from their neighbors, it qualitatively and quantitatively captures the features of configurationally hybridized graphene, which will be proved as follows. The predicted bandgaps of the 4 × 4 and 5 × 5 supercell systems by different DL algorithms are compared with the results by the DFT calculations (Fig. 3 ). Note that these data is obtained from the method of “learning from scratch” which suggests that the training and prediction are performed under the same graphene-h-BN hybrid systems. 44 For instance, the networks which are trained using the data from the 4 × 4 supercell systems are used to predict the systems with the same size but different configurations. The prediction accuracy is characterized by the relative error of the predicted bandgaps ( E ML ) to the DFT calculated bandgaps ( E DFT ), which is calculated as | E ML − E DFT |/ E DFT . As shown in Fig. 3a , all of three CNNs can predict the bandgaps of 4 × 4 supercell systems within 10% relative error for >90% cases. All of the predicted bandgaps for all cases have accuracy of >80%. In contrast, the prediction results from the SVM are deviated much more from the DFT benchmarks, showing >20% error for >50% cases. Figure 3b shows that the three CNNs exhibit strong direct linear correlation of ML predicted values and the DFT calculated values, while the SVM shows very weak correlation. The prediction accuracy of these CNNs for 5 × 5 supercell systems degrades a little (Fig. 3c ). But the VCN network shows the prediction accuracy of >90% for >90% cases, which is the best among all three CNNs. The CCN has the lowest with prediction accuracy of >90% for only ~50% cases. That is possible due to lack of training data for 5 × 5 supercell systems considering their much larger configuration space than 4 × 4 supercell systems. The ML predicted bandgaps still have strong linear correlation with the DFT calculated ones (Fig. 3d ). We believe that as the size of training datasets increases, the accuracy would be further improved. Similar to the prediction results shown for 4 × 4 supercell systems, when predicting bandgaps of 5 × 5 supercell systems the SVM shows poor performance (Fig. 3b, d ).

Prediction performance of four ML algorithms. a , c Error levels of ML predicted bandgaps for 4 × 4 supercell systems a and 5 × 5 supercell systems c . The cases used for prediction are arranged by percentage of predicted cases showing prediction errors of <10%, <20%, and >20%. b , d ML predicted bandgaps vs. DFT calculated values for 4 × 4 supercell systems b and 5 × 5 supercell systems d
In addition, other indicators of the prediction performance—the mean absolute error (MAE), root-mean-square error (RMSE) and explained variance ( R 2 )—are provided in Table 1 . Meanwhile, the fractional error, MAE F and RMSE F were also calculated, with their definition is shown in Supplementary Notes 5 . For the 4 × 4 supercell systems, all three CNNs show very low RMSE of ~0.1 eV for the predicted bandgaps. The corresponding fractional errors, RMSE F , for all three CNNs are ~6%. For the 5 × 5 supercell systems, the RMSE values slightly increase to 0.16 eV for the CCN, but it decreases to 0.09 eV and 0.10 eV when predicted by the VCN and RCN, further confirming the effectiveness of the VCN in predicting larger systems. The performance is also compared with different concentrations of BN pairs in the 5 × 5 supercell systems. Table S7 shows the statistics of prediction accuracy at three different levels of BN concentrations (<33 at%: “Low Concentration”; 33 at% ~ 66 at%: “Medium Concentration”; >66 at%: “High Concentration”). Compared with the results listed in Table 1 , the accuracy doesn’t change much, indicating the robustness of the CNN models in predicting the bandgaps of the structures with different BN concentrations.
The ultralow RMSE and RMSE F values show that these DL algorithms are more effective in predicting bandgaps of our system than other material systems, such as double perovskites 39 which show a RMSE of 0.36 eV or inorganic crystals which show a RMSE of 0.51 eV. 23 This advantage is even more compelling if considering that: (i) our performance is rigorously evaluated with the newly generated systems that don’t have any translational or symmetry equivalence with the training ones; (ii) the error maintains a low level when the relative size of the training data is significantly reduced for the 5 × 5 supercell systems. In contrast, the prediction accuracy from the SVM algorithm is lower, showing higher RMSEs of 0.33 and 0.51 eV for the 4 × 4 and 5 × 5 supercell systems, respectively. The R 2 is an indicator of correlation between the prediction and real values, which is considered as one of the most important metrics for evaluating the accuracy of the prediction models. Table 1 illustrates that the predicted bandgaps by all three CNNs have ~95% and >90% relevance to the values calculated by the DFT for the 4 × 4 and 5 × 5 supercell systems, respectively. Among them, the RCN shows the best prediction results. In contrast, the SVM has a near zero R 2 , indicating almost no relevance between the two. In summary, these results show that the CNNs are superior to non-CNN ML algorithms in predicting bandgaps of the configurationally hybridized graphene. People generally agree that the CNN methods perform better than non-CNN ones in terms of feature extraction for problems involving spatial structures. This advantage could be attributed to the convolution processing of data in CNNs. 45 , 46 , 47 The flattening process of SVM method could lost important spatial features which are deterministic to bandgaps. Therefore, our results presented in this work provide another example for the effectiveness of CNNs dealing with spatial problems in material science.
Transfer learning: training and prediction
As suggested from the prediction results shown in the 4 × 4 and 5 × 5 supercell systems, the prediction accuracy is decreased as the relative size of training data shrinks. Obtaining sufficient training data, such as bandgaps calculated by the DFT, can lead to unusually high cost especially as the system scales up. Such a problem imposes a major challenge in the application of ML to the materials science. To overcome this challenge, an emerging TL has been proposed. 48 To conceptually validate the effectiveness of the TL in predicting bandgaps of larger systems we leveraged relatively larger datasets generated from the 4 × 4 and 5 × 5 supercell systems to improve models trained on more limited datasets generated in the 6 × 6 supercell systems. To do that, we built TL frameworks based on the CCN, RCN, and VCN. These networks were trained with the datasets previously used for training the 4 × 4 and 5 × 5 supercell systems together with 7200 new data points generated from the 6 × 6 supercell systems. The TL procedures for all CNNs are similar to the ones shown in refs. 49 , 50 where all CNNs layers except the last FL are set at a learning rate 10% of the original learning rate. The last layer is re-normalized and trained with the new dataset. Its learning rate is set to 1% of the original CNN networks.
Figure 4a shows prediction errors of bandgaps for the 6 × 6 supercell systems by all three DL algorithms without the TL. Compared with the 4 × 4 and 5 × 5 systems they are significantly increased in all three categories of the prediction errors. The VCN shows >90% prediction accuracy for only ~40% cases. The RCN performs the best among all three ones with near 55% cases reaching > 90% accuracy. The CCN shows the lowest percentage (i.e., ~45%) of cases within 10% prediction error and has 15% of the cases with >20% error. Figure 4b shows that the correlation between the predicted bandgaps and the DFT calculated ones is weaker for all three CNNs compared with the smaller systems, due to the much smaller percentage of sample size (only 1 × 10 –7 of all possibilities). The prediction accuracy can be notably boosted after they are combined the TL (Fig. 4c ). The CCN with the TL performs the best, with >60% cases achieving >90% accuracy. The percentage of cases with >20% error reduces to <10% from 20%. It is a significant improvement considering that increasing prediction accuracy becomes more and more difficult after a certain point. With the TL, the RCN and VCN also shows improved prediction accuracy. The percentage of the cases that show >20% error decreases from 20 to 8% when using the RCN, while VCN decrease percentage of cases from 35 to 12%. The correlation between the predicted bandgaps and the DFT calculated ones becomes much stronger after using the TL methods (Fig. 4d ). The statistics of the predicted bandgaps by the different DL algorithms with and without the TL for the 6 × 6 supercell systems is shown in Table 2 . Overall, the TL boosts the prediction accuracy of all three DL algorithms in terms of reducing the MAE and RMSE. For instance, it helps to reduce the MAE of CCN from 0.13to 0.09 eV and RMSE from 0.16 to 0.12 eV. Their RMSE F values are also reduced. This accuracy is comparable to the prediction accuracy show in the 5 × 5 supercell systems with the same DL algorithms. They are the lowest among the values predicted by all three CNNs used in this work. This demonstration of applying the TL to predict bandgaps of the configurationally hybridized graphene of larger size would pave new route to mitigating barriers for the ML in solving challenges of data scarcity faced in the material fields.

Prediction performance of three DL algorithms a , b before and c , d after transfer learning for 6 × 6 supercell systems. a , c Error levels of ML predicted bandgaps before TL a and after TL c . The cases used for prediction are arranged by percentage of predicted cases showing prediction errors of <10%, <20%, and >20%. b , d ML predicted bandgaps vs. DFT calculated values before TL b and after TL d
In summary, we develop the DL models to predict the bandgaps of hybrids of graphene and h-BN with arbitrary supercell configurations. Three CNNs yield high prediction accuracy (>90%) for 4 × 4 and 5 × 5 supercell systems. The CNNs show superior performance to the non-CNN ML algorithms. The TL, leveraging the pre-trained network on small systems, boosts the prediction accuracy of three CNNs when predicting the bandgaps of large systems. The resulting MSE and RMSE of the predicted bandgaps of 6 × 6 systems by the CCN can be close to those of the 5 × 5 systems, but with a much smaller sampling ratio. Through this scientifically significant example, we have successfully illustrated the potential of artificial intelligence in studying 2D materials with various configurations, which would pave a new route to rational design of materials. Given the recent progress in atom-scale precise structures of 2D materials realized by bottom-up chemical synthesis, we anticipate that our methods could form a computational platform that enables to pre-screen candidates for experimental realization. Moreover, due to the extremely high computation cost from the DFT calculation for larger systems, 51 this platform could be useful to extrapolate the results obtained from smaller systems to larger systems with assistance of the TL algorithm. Upon the success of CNNs in present material systems, we envision that they could be extendable to 2D material systems with different number of layers considering that the CNN models are also effective in RGB images classification by preprocessing the images into multiple 2D numerical matrices. 52 In this case, the images can be encoded to 2D numerical matrices and then fed into the CNN models. Nevertheless, this hypothesis requires careful investigation due to layer-to-layer interaction resulted physical and chemical complexity. Nevertheless, we believe that current work will inspire researchers in 2D materials for further exploring this promising area.
DFT calculation
The ab initio DFT calculations were performed by QUANTUM ESPRESSO package. 53 We have successfully employed DFT calculations to investigate the nitrogen doping in graphene and to predict a class of novel two-dimensional carbon nitrides. 54 , 55 The ultra-soft projector-augmented wave (PAW) pseudopotential 56 was used to describe the interaction between the valence electrons and the ions. The Perdew-Burke-Ernzenhof (PBE) function was applied for the exchange-correlation energy. 57 The cutoff plane wave energy was set to 400 eV. The Monkhorst-Pack scheme 58 was applied to sample the Brillouin zone with a mesh grid from 12 × 12 × 1 in the k-point for all the systems. The graphene sheets were modeled as 2D matrixes. The matrix with all zero represents the intrinsic graphene, and the matrix with all one represents the intrinsic h-BN. The hybridized graphene and h-BN sheets were modeled by matrixes with 0 and 1 elements. The sizes of computed 2D sheets were 4 × 4, 5 × 5, and 6 × 6 supercells. The output training datasets were the bandgaps of randomly generated 2D graphene and BN hybrids with random concentration of BN pairs varying from 0 to 100 at% (Fig. S5 ). Specifically, the configurations were generated by random sampling. For example, for the 4 × 4 systems, we generated a set of random decimal numbers between 0 and2 16 . Then these decimal numbers were converted into binary numbers that were represented with 16 digits. These digits were further converted to 4 by 4 matrixes to represent samples showing various configurations.
Data availability
All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials . Additional data related to this paper may be requested from the authors.
Lu, K., Lu, L. & Suresh, S. Strengthening materials by engineering coherent internal boundaries at the nanoscale. Science 324 , 349–352 (2009).
Article CAS Google Scholar
Tian, Y. J. et al. Ultrahard nanotwinned cubic boron nitride. Nature 493 , 385–388 (2013).
Huang, Q. et al. Nanotwinned diamond with unprecedented hardness and stability. Nature 510 , 250–253 (2014).
Mamin, H. J. et al. Nanoscale nuclear magnetic resonance with a nitrogen-vacancy spin sensor. Science 339 , 557–560 (2013).
Grinolds, M. S. et al. Subnanometre resolution in three-dimensional magnetic resonance imaging of individual dark spins. Nat. Nanotechnol. 9 , 279–284 (2014).
Doi, Y. et al. Deterministic electrical charge-state initialization of single nitrogen-vacancy center in diamond. Phys. Rev. X 4 , 011057 (2014).
Google Scholar
Shinada, T., Okamoto, S., Kobayashi, T. & Ohdomari, I. Enhancing semiconductor device performance using ordered dopant arrays. Nature 437 , 1128–1131 (2005).
Kitchen, D., Richardella, A., Tang, J. M., Flatte, M. E. & Yazdani, A. Atom-by-atom substitution of Mn in GaAs and visualization of their hole-mediated interactions. Nature 442 , 436–439 (2006).
Koenraad, P. M. & Flatte, M. E. Single dopants in semiconductors. Nat. Mater. 10 , 91–100 (2011).
Kalff, F. E. et al. A kilobyte rewritable atomic memory. Nat. Nanotechnol. 11 , 926–929 (2016).
Grantab, R., Shenoy, V. B. & Ruoff, R. S. Anomalous strength characteristics of tilt grain boundaries in graphene. Science 330 , 946–948 (2010).
Shekhawat, A. & Ritchie, R. O. Toughness and strength of nanocrystalline graphene. Nat. Commun. 7 , 10546 (2016).
Ma, T. et al. Tailoring the thermal and electrical transport properties of graphene films by grain size engineering. Nat. Commun. 8 , 14486 (2017).
Tsen, A. W. et al. Tailoring electrical transport across grain boundaries in polycrystalline graphene. Science 336 , 1143–1146 (2012).
Fei, Z. et al. Electronic and plasmonic phenomena at graphene grain boundaries. Nat. Nanotechnol. 8 , 821–825 (2013).
Cruz-Silva, E., Barnett, Z. M., Sumpter, B. G. & Meunier, V. Structural, magnetic, and transport properties of substitutionally doped graphene nanoribbons from first principles. Phys. Rev. B 83 , 155445 (2011).
Article Google Scholar
Martins, T. B., Miwa, R. H., da Silva, A. J. R. & Fazzio, A. Electronic and transport properties of boron-doped graphene nanoribbons. Phys. Rev. Lett. 98 , 196803 (2007).
Kim, S. S., Kim, H. S., Kim, H. S. & Kim, Y. H. Conductance recovery and spin polarization in boron and nitrogen co-doped graphene nanoribbons. Carbon N. Y. 81 , 339–346 (2015).
Lherbier, A., Blase, X., Niquet, Y. M., Triozon, F. & Roche, S. Charge transport in chemically doped 2D graphene. Phys. Rev. Lett. 101 , 036808 (2008).
Yu, S. S., Zheng, W. T., Wen, Q. B. & Jiang, Q. First principle calculations of the electronic properties of nitrogen-doped carbon nanoribbons with zigzag edges. Carbon N. Y. 46 , 537–543 (2008).
Biel, B., Blase, X., Triozon, F. & Roche, S. Anomalous doping effects on charge transport in graphene nanoribbons. Phys. Rev. Lett. 102 , 096803 (2009).
Zheng, X. H., Rungger, I., Zeng, Z. & Sanvito, S. Effects induced by single and multiple dopants on the transport properties in zigzag-edged graphene nanoribbons. Phys. Rev. B 80 , 235426 (2009).
Isayev, O. et al. Universal fragment descriptors for predicting properties of inorganic crystals. Nat. Commun. 8 , 15679 (2017).
Raccuglia, P. et al. Machine-learning-assisted materials discovery using failed experiments. Nature 533 , 73–76 (2016).
Xue, D. Z. et al. Accelerated search for BaTiO3-based piezoelectrics with vertical morphotropic phase boundary using Bayesian learning. Proc. Natl Acad. Sci. USA 113 , 13301–13306 (2016).
Xue, D. Z. et al. Accelerated search for materials with targeted properties by adaptive design. Nat. Commun. 7 , 11241 (2016).
Nikolaev, P. et al. Autonomy in materials research: a case study in carbon nanotube growth. npj Comput. Mater. 2 , 16031 (2016).
Tabor, D. P. et al. Accelerating the discovery of materials for clean energy in the era of smart automation, Nature Reviews. Materials 3 , 5–20 (2018).
CAS Google Scholar
Agrawal, A. & Choudhary, A. Perspective: materials informatics and big data: Realization of the “fourth paradigm” of science in materials science. APL Mater. 4 , 053208 (2016).
LeCun, Y., Bengio, Y. & Hinton, G. Deep learning. Nature 521 , 436–444 (2015).
Gibney, E. Google AI algorithm masters ancient game of Go. Nature 529 , 445–446 (2016).
Carrasquilla, J. & Melko, R. G. Machine learning phases of matter. Nat. Phys. 13 , 431–434 (2017).
Ziletti, A., Kumar, D., Scheffler, M. & Ghiringhelli, L. M. Insightful classification of crystal structures using deep learning. Nat. Commun. 9 , 2775 (2018).
Cortes, C. & Vapnik, V. Support-vector networks. Mach. Learn. 20 , 273–297 (1995).
Kawai, S. et al. Atomically controlled substitutional boron-doping of graphene nanoribbons. Nat. Commun. 6 , 8098 (2015).
Nguyen, G. D. et al. Atomically precise graphene nanoribbon heterojunctions from a single molecular precursor. Nat. Nanotechnol. 12 , 1077–1082 (2017).
Kawai, S. et al. Multiple heteroatom substitution to graphene nanoribbon. Sci. Adv. 4 , 7181 (2018).
Jin, C., Lin, F., Suenaga, K. & Iijima, S. Fabrication of a freestanding boron nitride single layer and its defect assignments. Phys. Rev. Lett. 102 , 195505 (2009).
Pilania, G. et al. Machine learning bandgaps of double perovskites. Sci. Rep. 6 , 19375 (2016).
Simonyan K., Zisserman A. Very deep convolutional networks for large-scale image recognition, arXiv, arXiv : 1409, 1556 [cs.CV], (2014).
Russakovsky, O. et al. Imagenet large scale visual recognition challenge. Int. J. Comput. Vision. 115 , 211–252 (2015).
Szegedy, C. et al. Going deeper with convolutions. Proc. IEEE Conf. Comput. Vision. Pattern Recognit. 2015 , 1–9 (2015).
Huang, G., Liu, Z., Van Der Maaten, L. & Weinberger, K. Q. Densely connected convolutional networks. CVPR 2017 , 3 (2017).
Pan, S. J. & Yang, Q. A. A survey on transfer learning. IEEE Trans. Knowl. Data Eng. 22 , 1345–1359 (2010).
Sharif Razavian, A., Azizpour, H., Sullivan, J. & Carlsson, S. CNN features off-the-shelf: an astounding baseline for recognition. Proc. IEEE Conf. Comput. Vision. Pattern Recognit. Workshops 2014 , 806–813 (2014).
Liu, Z., Hu, J., Weng, L. & Yang, Y. Rotated region based CNN for ship detection. Image Processing (ICIP). 2017 IEEE Int. Conf. 2017 , 900–904 (2017).
Yin, W., Kann, K., Yu, M. & Schütze, H. Comparative study of cnn and rnn for natural language processing. arXiv Prepr. arXiv 1702 , 01923 (2017).
Hutchinson, M. L. et al. Overcoming data scarcity with transfer learning. arXiv, arXiv 1711 , 05099 (2017).
Girshick, R., Donahue, J., Darrell, T. & Malik, J. Region-based convolutional networks for accurate object detection and segmentation. IEEE. Trans. Pattern Anal. Mach. Intell. 38 , 142–158 (2016).
Hoo-Chang, S. et al. Deep convolutional neural networks for computer-aided detection: CNN architectures, dataset characteristics and transfer learning. IEEE Trans. Med. Imaging 35 , 1285 (2016).
Ci, L. et al. Atomic layers of hybridized boron nitride and graphene domains. Nat. Mater. 9 , 430 (2010).
CireşAn, D., Meier, U., Masci, J. & Schmidhuber, J. Multi-column deep neural network for traffic sign classification. Neural Netw. 32 , 333–338 (2012).
Giannozzi, P. et al. QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials. J. Phys.: Condens. Matter 21 , 395502 (2009).
Dong, Y., Gahl, M. T., Zhang, C. & Lin, J. Computational study of precision nitrogen doping on graphene nanoribbon edges. Nanotechnology 28 , 505602 (2017).
Dong, Y., Zhang, C., Meng, M., Melinda, G. & Lin, J. Novel two-dimensional diamond like carbon nitrides with extraordinary elasticity and thermal conductivity. Carbon 138 , 319–324 (2018).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59 , 1758 (1999).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77 , 3865 (1996).
Monkhorst, H. J. & Pack, J. D. Special points for Brillouin-zone integrations. Phys. Rev. B 13 , 5188 (1976).
Download references
Acknowledgements
J.L. acknowledges financial support from University of Missouri-Columbia start-up fund, NASA Missouri Space Consortium (Project: 00049784), Unite States Department of Agriculture (Award number: 2018-67017-27880). This material is based upon work partially supported by the Department of Energy National Energy Technology Laboratory under Award Number DE-FE0031645. J.C. acknowledges National Science Foundation (Award numbers: DBI1759934 and IIS1763246). The computations were performed on the HPC resources at the University of Missouri Bioinformatics Consortium (UMBC), supported in part by NSF (award number: 1429294).
Author information
These authors contributed equally: Yuan Dong, Chuhan Wu
Authors and Affiliations
Department of Mechanical & Aerospace Engineering, University of Missouri, Columbia, MO, 65211, USA
Yuan Dong, Chi Zhang & Jian Lin
Department of Electrical Engineering & Computer Science, University of Missouri, Columbia, MO, 65211, USA
Chuhan Wu, Yingda Liu & Jianlin Cheng
You can also search for this author in PubMed Google Scholar
Contributions
J.L. conceived the project. Y.D. designed material descriptive system, performed the DFT calculations, and did the data analysis. C.Z. assisted Y.D. in DFT calculation and data analysis. J.C. designed the deep learning methods. C.W. built the CNNs and performed the training and prediction assisted by Y. Liu. J.L. and J.C. supervised the project. Y.D. and C.W. contributed equally to this work. All authors contributed to the discussions and writing of the manuscript.
Corresponding authors
Correspondence to Jianlin Cheng or Jian Lin .
Ethics declarations
Competing interests.
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplemental material, rights and permissions.
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/ .
Reprints and permissions
About this article
Cite this article.
Dong, Y., Wu, C., Zhang, C. et al. Bandgap prediction by deep learning in configurationally hybridized graphene and boron nitride. npj Comput Mater 5 , 26 (2019). https://doi.org/10.1038/s41524-019-0165-4
Download citation
Received : 08 October 2018
Accepted : 06 February 2019
Published : 26 February 2019
DOI : https://doi.org/10.1038/s41524-019-0165-4
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
This article is cited by
Deep learning in two-dimensional materials: characterization, prediction, and design.
- Xinqin Meng
- Chengbing Qin
- Suotang Jia
Frontiers of Physics (2024)
Investigating the effect of textural properties on CO2 adsorption in porous carbons via deep neural networks using various training algorithms
- Pardis Mehrmohammadi
- Ahad Ghaemi
Scientific Reports (2023)
Faux-Data Injection Optimization for Accelerating Data-Driven Discovery of Materials
- Abdul Wahab Ziaullah
- Sanjay Chawla
- Fedwa El-Mellouhi
Integrating Materials and Manufacturing Innovation (2023)
Graph neural network for Hamiltonian-based material property prediction
- Haibin Ling
Neural Computing and Applications (2022)
Machine learning-based solubility prediction and methodology evaluation of active pharmaceutical ingredients in industrial crystallization
- Zhenguo Gao
Frontiers of Chemical Science and Engineering (2022)
Quick links
- Explore articles by subject
- Guide to authors
- Editorial policies
Sign up for the Nature Briefing newsletter — what matters in science, free to your inbox daily.

IMAGES
VIDEO
COMMENTS
A large dataset has been collated for bandgap prediction of many perovskite types. • An existing GNN model has been trained on the dataset and model has been interpreted. • Some key features for further development of ML models are also highlighted. • A pipeline has been created for easily predicting bandgaps for inorganic perovskites.
The contributions we make in our paper are summarized here: 1) A large dataset has been collated for bandgap prediction of perovskites. A dataset description has also been provided. 2) An existing GNN model has been trained on this dataset and experiments have been performed to interpret the model.
DOI: 10.1016/J.COMMATSCI.2021.110530 Corpus ID: 236237757; Graph representational learning for bandgap prediction in varied perovskite crystals @article{Omprakash2021GraphRL, title={Graph representational learning for bandgap prediction in varied perovskite crystals}, author={P. Omprakash and Bharadwaj Manikandan and Ankit Sandeep and Romit Shrivastava and Viswesh P. and Devadas Bhat ...
In 2021, Omprakash et al. 169 predicted the energy band gap based on a large database containing 27 400 kinds of perovskite. The model, based on the Materials Graph Network containing the periodic ...
In this project, the computational efficiency of Graph Neural Networks (GNNs) has been leveraged to help accelerate the discovery of new perovskite compounds (ABX3) with favourable properties. The GNN used in the model helps predict with a high degree of accuracy the bandgap of perovskites in a few milliseconds and given its high ...
The inset indicates the zoomed-in histogram for for the 310 oxide perovskites with a predicted wide band gap probability between 0.9 and 1. c Histogram of predicted band gaps for the 310 oxide ...
Year. Graph Representational Learning for bandgap prediction in varied perovskite crystals. DBP Pravan Omprakash, M Bharadwaj, R Srivastava, A Sandeep, Viswesh P. Computational Material Science 196 (August 2021), 110530. , 2021. 19. 2021. A Review of 2D Perovskites and Carbon-Based Nanomaterials for Applications in Solar Cells and Photodetectors.
The band gap is an important parameter that determines light-harvesting capability of perovskite materials. It governs the performance of various optoelectronic devices such as solar cells, light-emitting diodes, and photodetectors. For perovskites of a formula ABX3 having a non-zero band gap, we study nonlinear mappings between the band gap and properties of constituent elements (e.g ...
Graph representational learning for bandgap prediction in varied perovskite crystals [Display omitted] •A large dataset has been collated for bandgap prediction of many perovskite types.•An existing GNN model has been trained on the dataset and model has been interpreted.•Some key features for further development of ML models are also highlighted.•A pipeline has been created for eas...
The role of anion and cations and their impact on optoelectronic and photovoltaic properties is probed. A machine learning (ML) approach to predict the bandgap and power conversion efficiency (PCE) using eight different perovskites compositions is reported. The predicted solar cell parameters validate the experimental data.
Graph Representational Learning for bandgap prediction in varied perovskite crystals. P Omprakash, B M, R Srivastava, A Sandeep, V P, ... Computational Material Science, 2021. 20: 2021: Energy Storage through Graphite Intercalation Compounds. V Gopalakrishnan, A Sundararajan, P Omprakash, DB Panemangalore.
Graph representational learning for bandgap prediction in varied perovskite crystals Computational Materials Science ( IF 3.3) Pub Date : 2021-05-06, DOI: 10.1016/j.commatsci.2021.110530
Here, we combine positive-unlabeled learning 26, domain-specific learning 55, and transfer learning 56 to develop the synthesizability prediction model of perovskites with a high practical ...
Halide perovskite materials have broad prospects for applications in various fields such as solar cells, LED devices, photodetectors, fluorescence labeling, bioimaging, and photocatalysis due to their bandgap characteristics. This study compiled experimental data from the published literature and utilized the excellent predictive capabilities, low overfitting risk, and strong robustness of ...
In this project, the computational efficiency of Graph Neural Networks (GNNs) has been leveraged to help accelerate the discovery of new perovskite compounds (ABX3) with favourable properties. The GNN used in the model helps predict with a high degree of accuracy the bandgap of perovskites in a few milliseconds and given its high ...
Deviations from this value can induce different crystal symmetries like orthorhombic or tetragonal, affecting the material properties such as electronic conductivity, bandgap structure, and stability. ... our work takes a more comprehensive approach, combining the prediction of crystal structure, band gap, and formation energy to provide a more ...
select article Graph representational learning for bandgap prediction in varied perovskite crystals. ... Research article Full text access Graph representational learning for bandgap prediction in varied perovskite crystals. Pravan Omprakash, Bharadwaj Manikandan, Ankit Sandeep, Romit Shrivastava, ... Devadas Bhat Panemangalore. Article 110530 ...
Graph representational learning for bandgap prediction in varied perovskite crystals. ... A hierarchical screening process is implemented in which two cross-validated and predictive machine learning models for band gap classification and regression, trained using exhaustive datasets that span 68 elements of the periodic table, are applied ...
DOI: 10.1016/j.physleta.2021.127800 Corpus ID: 243479672; Bandgap prediction of metal halide perovskites using regression machine learning models @article{Vakharia2021BandgapPO, title={Bandgap prediction of metal halide perovskites using regression machine learning models}, author={Vinay Vakharia and Ivano Eligio Castelli and Keval Bhavsar and Ankur Solanki}, journal={Physics Letters A}, year ...
The prediction results of the optimal ΔH and ΔL showed that when PCE was high, it was low and high ΔH and ΔL values were suitable for low and high bandgap halide perovskite, respectively. In 2018, Takahashi et al. [ 59 ] calculated the data of 15,000 halide perovskite materials with the first principle and used the data to find unknown ...
Application of machine learning to perovskite material properties and candidate screening. ... crystal growth for high-performance all-inorganic perovskite solar cells. Energy Environ. Sci. ... Graph representational learning for bandgap prediction in varied perovskite crystals. Computational Materials Science, Volume 196, 2021, Article 110530 ...
Graph representational learning for bandgap prediction in varied perovskite crystals. ... A hierarchical screening process is implemented in which two cross-validated and predictive machine learning models for band gap classification and regression, trained using exhaustive datasets that span 68 elements of the periodic table, are applied ...
Graph representational learning for bandgap prediction in varied perovskite crystals. Computational Materials Science 2021-08 | Journal article DOI: 10.1016/j.commatsci.2021.110530 Part of ISSN: 0927-0256 Show more detail. Source: ...
Note that these data is obtained from the method of "learning from scratch" which suggests that the training and prediction are performed under the same graphene-h-BN hybrid systems. 44 For ...