- Skip to primary navigation
- Skip to main content
- Skip to primary sidebar
- Skip to footer
- Image & Use Policy
- Translations
UC MUSEUM OF PALEONTOLOGY

Understanding Evolution
Your one-stop source for information on evolution
- ES en Español

The genetic variation in modern human populations has been critically shaped by gene flow. For example, by sequencing ancient DNA, researchers have reconstructed the entire Neanderthal genome – and they’ve found that many snippets of these archaic sequences live on in modern humans. It’s clear that ancient humans and Neanderthals interbred, and that this gene flow introduced new genetic variation to the human population. Furthermore, this ancient gene flow seems to affect who we are today. Neanderthal gene versions have been linked to immune functions, metabolic functions (e.g., affecting one’s risk of developing diabetes), and even skin color.
- More Details
- Evo Examples
Read more about the details of gene flow .
Read more about the results of gene flow from ancient human populations into our ancestors in these news briefs:
- Genes from our extinct relatives live on in modern humans
- The deep roots of diabetes
Reviewed and updated June, 2020.
The causes of mutations
Sex and genetic shuffling
Subscribe to our newsletter
- Teaching resource database
- Correcting misconceptions
- Conceptual framework and NGSS alignment
- Image and use policy
- Evo in the News
- The Tree Room
- Browse learning resources
- History & Society
- Science & Tech
- Biographies
- Animals & Nature
- Geography & Travel
- Arts & Culture
- Games & Quizzes
- On This Day
- One Good Fact
- New Articles
- Lifestyles & Social Issues
- Philosophy & Religion
- Politics, Law & Government
- World History
- Health & Medicine
- Browse Biographies
- Birds, Reptiles & Other Vertebrates
- Bugs, Mollusks & Other Invertebrates
- Environment
- Fossils & Geologic Time
- Entertainment & Pop Culture
- Sports & Recreation
- Visual Arts
- Demystified
- Image Galleries
- Infographics
- Top Questions
- Britannica Kids
- Saving Earth
- Space Next 50
- Student Center
- When did science begin?
- Where was science invented?
- Why is biology important?

Our editors will review what you’ve submitted and determine whether to revise the article.
- National Center for Biotechnology Information - PubMed Central - When gene flow really matters: gene flow in applied evolutionary biology
gene flow , the introduction of genetic material (by interbreeding ) from one population of a species to another, thereby changing the composition of the gene pool of the receiving population. The introduction of new alleles through gene flow increases variability within the population and makes possible new combinations of traits. In humans gene flow usually comes about through the actual migration of human populations, either voluntary or forced.
Although gene flow does not change allele frequencies for a species as a whole, it can alter allele frequencies in local populations. In the case of migration, the greater the difference in allele frequencies between the resident and the migrant individuals, and the larger the number of migrants, the greater the effect the migrants have in changing the genetic constitution of the resident population.
- Open access
- Published: 10 November 2022
Gene flow and introgression are pervasive forces shaping the evolution of bacterial species
- Awa Diop 1 ,
- Ellis L. Torrance 1 ,
- Caroline M. Stott 1 &
- Louis-Marie Bobay ORCID: orcid.org/0000-0002-0438-545X 1
Genome Biology volume 23 , Article number: 239 ( 2022 ) Cite this article
4955 Accesses
9 Citations
31 Altmetric
Metrics details
Although originally thought to evolve clonally, studies have revealed that most bacteria exchange DNA. However, it remains unclear to what extent gene flow shapes the evolution of bacterial genomes and maintains the cohesion of species.
Here, we analyze the patterns of gene flow within and between >2600 bacterial species. Our results show that fewer than 10% of bacterial species are truly clonal, indicating that purely asexual species are rare in nature. We further demonstrate that the taxonomic criterion of ~95% genome sequence identity routinely used to define bacterial species does not accurately represent a level of divergence that imposes an effective barrier to gene flow across bacterial species. Interruption of gene flow can occur at various sequence identities across lineages, generally from 90 to 98% genome identity. This likely explains why a ~95% genome sequence identity threshold has empirically been judged as a good approximation to define bacterial species. Our results support a universal mechanism where the availability of identical genomic DNA segments required to initiate homologous recombination is the primary determinant of gene flow and species boundaries in bacteria. We show that these barriers of gene flow remain porous since many distinct species maintain some level of gene flow, similar to introgression in sexual organisms.
Conclusions
Overall, bacterial evolution and speciation are likely shaped by similar forces driving the evolution of sexual organisms. Our findings support a model where the interruption of gene flow—although not necessarily the initial cause of speciation—leads to the establishment of permanent and irreversible species borders.
Gene flow is a key evolutionary process upon which theoretical frameworks of speciation have been primarily founded in sexual organisms. In contrast, bacteria reproduce asexually, and developing a theoretical framework to establish a definition of bacterial species has proven difficult; and some have even questioned the existence of prokaryotic species [ 1 , 2 ]. As a result, most studies rely on operational definitions of bacterial species, which are often based on arbitrary sequence thresholds [ 3 , 4 ]. Albeit convenient, these definitions impede our ability to fully understand the evolution and dynamics of bacteria. Some bacteria can engage in gene flow via homologous recombination [ 5 ] and this observation has led a growing number of researchers to suggest that bacterial species and speciation might be best defined using the same evolutionary theory developed for sexual organisms [ 6 , 7 , 8 , 9 , 10 , 11 ]; the biological species concept (BSC) [ 12 , 13 , 14 ]. It has been long established that even distantly related bacteria can occasionally exchange genes through horizontal gene transfers (HGT), but this process usually involves accessory genes that are not part of the genomic backbone—the core genome—of the species. The core genome, representing the most functionally important set of genes, is thought to evolve primarily vertically [ 15 ] and thus, is the focus of most efforts to understand the population structure and evolution of bacteria. Despite many years of work, the number of bacterial species engaging in gene flow and the limits of gene flow between populations and species remain poorly defined. Therefore, although bacteria are often presumed to evolve clonally, the prevalence of truly clonal species remains unknown [ 5 ]. Additionally, some bacterial species appear "fuzzy" as ongoing gene flow can be maintained between the core genomes of rather distant species and these processes might be analogous to the patterns of introgression frequently observed in sexual organisms [ 16 , 17 , 18 ]. Understanding the impact of DNA flux and developing a theory-anchored bacterial species concept remains a fundamental gap in evolutionary biology and microbiology. Key to this problem is our ability to recognize (i) which populations do and do not engage in gene flow, (ii) which bacterial species are truly clonal, and (iii) to what extent distant species can engage in gene flow.
Here we analyzed the patterns of gene flow within and between species across >2600 bacterial species and >30,000 genomes. We identified which of these bacterial species are truly clonal, classified bacteria into biological species by redefining species boundaries based on gene flow, and analyzed the patterns of gene flow between species (i.e., introgression). We show that very few bacterial species (2.6%) are unambiguously clonal, suggesting that truly asexual lineages are extremely rare across the Tree of Life. Our results also indicate that introgression is frequent in bacteria and that genomic divergence is the main factor determining the frequency of introgression events between species. Overall, our findings support a universal model of sexual isolation in bacteria where the decreasing frequency of identical DNA segments—which are required to initiate homologous recombination—appears to be the primary determinant of the interruption of gene flow. It further suggests that this mechanism could lead to the establishment of permanent species barriers between bacterial populations.
Results and Discussion
Very few bacteria are truly clonal.
Bacteria reproduce asexually but are known to frequently engage in homologous recombination. Despite many studies on recombination, it remains unclear how many and which bacterial species can be considered truly clonal in nature. Here we addressed this question by analyzing signals of recombination across a large set of bacterial species. We first reclassified all bacterial species for which at least 15 complete genomes have been sequenced using sequence identity thresholds commonly used to define bacterial species. To do this, we used the Average Nucleotide Identity (ANI) among the core genes with a cutoff value of 94% core-genome identity to reclassify named bacterial species into ANI-redefined species (Fig. 1 a, Additional file 1 : Table S1) [ 19 ]. The species containing less than 15 genomes after redefinition were excluded and this step yielded a total of 227 ANI-redefined species. To identify clonal species (Fig. 1 b), we first analyzed the patterns of homoplasic alleles ( h ) relative to non-homoplasic alleles ( m ). Homoplasic alleles are those whose distribution is incompatible with a scenario of vertical inheritance from a single common ancestor. They are likely the result of recombination events, but some can also accumulate due to independent convergent mutations. Thus, clonal species are expected to present low h/m ratios, but the exact number of homoplasic alleles found depends on the rate of substitutions, the age since divergence, and substitution biases. Therefore, we simulated the evolution of each species ( n =227) under a model of purely clonal evolution with substitution parameters that closely mimic the dataset for each species. We then estimated the h/m ratio for each species and the corresponding set of simulated genomes. Truly clonal species are expected to display h/m ratios similar to the simulated genomes, whereas recombining species should show higher h/m ratios when compared to the simulations (Additional file 2 : Fig. S1a). Next, we used the patterns of Linkage disequilibrium (LD) to infer clonality. In recombining genomes, LD (measured by r 2 ) decreases relative to genomic distances between the two loci (Additional file 2 : Figs. S1b, d). Due to the absence of recombination, clonal species should not exhibit a significant decrease between r 2 and genomic distances (Additional file 2 : Fig. S1b-c).

Approach used to reclassify bacterial species. The set of complete genomes for each species containing ≥15 genomes was analyzed and used as reference species. a Each reference species was redefined based on sequence identity thresholds using the ANI of core genes metric, i.e., the pairwise identity score computed on the core genome of each species. Genomes were considered as part of the same species when sharing at least 94% ANI of core genes. b Clonal species were inferred based on simulations without gene flow and based on Linkage Disequilibrium analysis. Reference species inferred as clonal by at least one of these methods were excluded from the next steps of the analysis. c Reference species were redefined into BSC species based on gene flow: genomes that showed a significant reduction in gene flow with the rest of the population were excluded from the reference species. d The redefined reference species based on steps B and C were tested for gene flow against candidate species. Candidate species were selected as species related to the reference species based on either taxonomic nomenclature (i.e., same genus name) or based on sequence relatedness. In this theoretical example, four candidate species were tested for gene flow against the reference species. One candidate species was found to engage in gene flow with the reference species and those were reclassified as the same species (species A). The three other candidate species were not found to engage in gene flow with the reference species and their original classification was kept (species B, C, and D)
Overall, we inferred that only 9.7% and 5.7% of species were clonal based on h/m ratios and LD respectively, with 2.6% of clonal species being inferred by both methods and a total of 12.8% inferred by at least one method (Additional file 2 : Fig. S2). Importantly, we noted that the species that we predicted as clonal did not present significantly lower amounts of polymorphisms (Additional file 2 : Fig. S2c) relative to the non-clonal ones, suggesting that lower statistical power and the accuracy of parameter estimation did not substantially bias our analysis. Our results indicate that most bacterial species display clear signs of recombination and as little as 2.6% appear truly clonal (Additional file 3 : Table S2). Species inferred as purely clonal were often endosymbionts from the genera Chlamydia , Brucella, and Bordetella , which have previously been defined as clonal [ 20 , 21 , 22 ]. However, not all endosymbiont species were found to be systematically clonal (Additional file 3 : Table S2).
Previous works have noted that adaptive evolution can sometimes lead to the presence of convergent mutations [ 23 , 24 , 25 ]. Although these observations were usually made in genes facing very strong selective pressures (e.g., antibiotic resistance genes) and/or during experimental evolution studies [ 23 ] we tested whether adaptive evolution could be responsible for the accumulation of a substantial fraction of the homoplasies detected in bacterial core genomes. We reasoned that if adaptive evolution were responsible for the presence of many homoplasies, those would be predominantly observed at non-synonymous sites where opportunities for positive selection are much higher than at synonymous sites. Indeed, synonymous sites are evolving under weak selective pressures which are primarily attributed to codon usage and nucleotide composition [ 26 ] and are typically acting at the scale of the entire gene and not at a particular site. For each species, we estimated the fractions of homoplasic and non-homoplasic alleles that were found at synonymous and non-synonymous positions across core genomes. Our results indicate that >50% homoplasic alleles are found at synonymous sites (Additional file 2 : Fig. S3), and, importantly, we observed that homoplasic alleles were not found more frequently at non-synonymous sites when compared to non-homoplasic alleles. In fact, the opposite trend was observed: homoplasic alleles are more frequently synonymous when compared to non-homoplasic alleles for ~95% of species (Additional file 2 : Fig. S3). Therefore, since the distribution of homoplasic alleles is not biased toward non-synonymous positions, adaptive selection is unlikely to explain the presence of homoplasies in the core genome of these species. Although this analysis does not completely exclude the possibility that some homoplasies are the result of positive selection, it should be noted that the homoplasies that we inferred are pervasive across bacterial core genomes with an average of 65,670 homoplasies per reference species (Additional file 3 : Table S2). This represents an average of 35 homoplasies per core gene across reference species (Additional file 3 : Table S2), indicating that our signal of gene flow is not driven by a few isolated genes. This strongly supports the view that positive selection plays a negligible role in the observed patterns of homoplasies.
Our results show that bacteria—like eukaryotes—present very few truly asexual lineages. As hypothesized for eukaryotes [ 27 ] truly asexual species of bacteria could be short-lived and therefore rare in nature (e.g., due to Muller’s ratchet). In contrast, Buchnera aphidicola and other insect endosymbionts are well-studied cases of strictly clonal bacteria that have co-evolved within their insect hosts for millions of years [ 28 ] but those were not included in this study due to the scarcity of genomes available and the parameters used to define initial species boundaries. It is likely that these ancient clonal lineages managed to escape extinction—i.e., to prolong their existence—by making themselves indispensable for their hosts by synthetizing several essential amino acids absent from the diet of sap-feeding insects, although these bacteria can eventually be replaced by new symbionts [ 29 ]. In summary, clonal bacteria are likely short-lived in nature, but some symbiotic species might be able to avoid extinction for substantially longer periods of time by evolving into organelle-like entities.
Gene flow delineates biological species boundaries
We reclassified bacteria based on gene flow by comparing patterns of recombination within and between species. We have previously developed an approach that was shown to identify the presence and the interruption of gene flow in bacteria as well as in sexual organisms [ 9 , 30 ]. First, we reclassified our set of non-clonal species ( n =198) based on gene flow (Fig. 1 c), as in [ 9 , 30 ], by excluding genomes showing a significant drop in h/m ratio ( within species redefinition). We redefined ~25% of species, which contained some non-recombining genomes and those were excluded from the species, yielding a total of 197 biological species (BSC species) (Additional file 1 : Table S1). The redefined species were then used as the set of "reference species" against which other species were compared ( between species comparison). Briefly, we selected every candidate species related to at least one of the reference species of our dataset and analyzed patterns of gene flow against each of the related reference species (Fig. 1 d). The candidate species were selected by downloading all named species within the same genus of the reference species available on NCBI, and also closely related species from other genera (see the Methods section). Following this process, we analyzed a total of 2446 candidate species by building the set of core genes shared between each candidate species and each reference species. Each pair of reference/candidate species was analyzed for gene flow by computing h/m ratios and conducting a resampling analysis to test the robustness of our metric. Although gene flow varies to some extent along a species’ core genome, these fluctuations are rather modest [ 31 , 32 ] and our sets of core genomes were typically composed of hundreds to thousands of genes for each pair of reference/candidate species (Additional file 4 : Table S3), thereby ensuring robust genome-wide estimates. A fraction of homoplasies can be introduced by convergent mutations and those are expected to increase in frequency as substitutions accumulate during genome divergence. Therefore, for all pairs of reference/candidate species, simulated genomes were evolved in silico without recombination and with similar characteristics and divergence rates as the candidate species relative to the reference species. Each simulated candidate species was then used to compute the ratio h/m 0 which represents the h/m ratio expected to result from convergent mutations alone since the divergence from the last common ancestor shared with the reference species. Using this procedure, we computed the adjusted ratio h/m norm for each pair of reference/candidate species. This metric corresponds to the h/m ratio adjusted for the amount of homoplasies expected to result from convergent mutations ( h/m 0 ) and the amount of homoplasies estimated in the reference species alone ( h/m ref ). If the candidate and the reference species are freely engaging in gene flow, the h/m ratio should be very similar to h/m ref ( h/m norm = 1). In contrast, if the two species do not engage in gene flow at all, the h/m ratio should be very similar to h/m 0 ( h/m norm = 0). As expected, we observed that most candidate species do not engage in gene flow with the corresponding reference species (Fig. 2 a, Additional file 2 : Fig. S4 and Additional file 4 : Table S3) as most h/m norm values were close to 0. However, we identified that 11.3% of all candidate species unambiguously engage in gene flow with other species ( h/m is not significantly lower than h/m ref and is significantly higher than h/m 0 ), indicating that these species pairs can be considered a single BSC species. Among reclassified species appeared well-documented cases of ambivalent taxonomy such as Escherichia coli and Shigella , which have been suggested to be classified into the same species based on sequence identity thresholds and phylogenetic analyses [ 33 , 34 , 35 ]. Moreover, we inferred that the species of the Burkholderia cepacia complex (Bcc)—a group of ubiquitous opportunistic pathogens which has undergone frequent taxonomic changes [ 36 , 37 ]—constitute a single biological species. The species of the Bcc complex are all closely related, with at least 90% ANI of core genes. Overall, the vast majority of all redefined BSC species in our dataset were closely related, i.e., typically >90% ANI of core genes (Fig. 2 b, c, Additional file 4 : Table S3). A few redefined species were distantly related (<80%) but displayed weak signals of gene flow (Additional file 2 : Fig. S5).

Patterns of gene flow across species. a Distribution of adjusted h/m ratios between candidate and reference species. The adjusted ratio of homoplasic to non-homoplasic alleles h/m norm was computed to quantify gene flow. h/m norm was computed by adjusting the values of h/m on the candidate and reference species ( h/m cand ) relative to h/m of the reference species alone ( h/m ref ) and relative to the h/m ratio expected to result from convergent mutations alone ( h/m 0 ). The adjusted ratio h/m norm is expected to be null when h/m cand is equal to h/m 0 ( h/m cand = h/m 0 ). The adjusted ratio h/m norm is expected to be near unity when h/m cand is similar to h/m ref ( h/m cand = h/m ref ). Only values ranging between 0 and 1 were represented (several datapoints exceeded 1, n =191, 1.6%). b Distribution of maximum sequence identities (ANI of core genes) between candidate and reference species that were reclassified as part of the same species based on gene flow. c Distribution of maximum sequence identities (ANI of core genes) between candidate and reference species that were not reclassified as part of the same species based on gene flow
Pervasive introgression across bacteria
We reclassified several species into a single BSC species based on the presence/absence of gene flow. However, the analysis of homoplasies revealed that many species pairs present intermediate levels of h/m ratios (Fig. 2 a) which are neither compatible with similar levels of gene flow observed in the reference species nor with the complete absence of gene flow. This raises the following question: Although two groups of genomes may be classified into distinct species due to lowered exchanges of gene flow, to what extent can they maintain some level of gene flow? Measuring gene flow based on h/m ratios cannot unequivocally address this question. Indeed, our h/m metric can retain some of the signal of gene flow even though gene flow might no longer be ongoing between two sets of genomes. Namely, intermediate levels of gene flow, as measured by h/m ratios, might not represent reduced ongoing gene flow between two species, but rather could reflect a complete—but recent—interruption of gene flow. To address this question, we derived a metric to quantify gene flow between candidate and reference species that were not reclassified as part of the same species and we named this metric introgression score (S i ) . For each of the 13,437 pairs of candidate/reference species (Additional file 4 : Table S3), we ran a 100bp scanning window along the core genome concatenate. Each 100bp fragment was defined as a transfer between the two species if at least one genome of the reference species was more similar to the candidate species than another genome of the reference species. The introgression score was then defined as the percent of the core genome that has been exchanged between the candidate and the reference species. Quantifying gene flow with this approach revealed a clear positive correlation between S i and h/m (Additional file 2 : Fig. S6), but many candidate/reference species pairs exhibited a lower introgression score relative to h/m , as predicted based on the fact that h/m can retain older signals of gene flow. Most candidate/reference species pairs were inferred to present rather modest amounts of introgressed DNA in their core genome (Fig. 3 ). The amount of gene flow inferred by S i varied widely across candidate/reference species pairs, but on average, 5.2% of the core genome was found to be exchanged between pairs of species with 46.3% of all species pairs presenting >1% of introgressed DNA (Fig. 3 , Additional file 4 : Table S3).

Levels of introgression across species. The graph represents the inferred fraction of candidate/reference species pairs presenting over 1%, 5%, and 10% of introgressed DNA in their core genome, respectively
Interestingly, we observed uneven levels of introgression across lineages. Species of the classes Alpha-Proteobacteria and Beta-Proteobacteria displayed some of the highest levels of introgression, whereas species of the Spirochaetia , Clostridia , and Chlamydiia showed very little signs of introgression (Additional file 2 : Fig. S7). Several species in our dataset were previously found to have exchanged DNA through introgression: Campylobacter jejuni and C. coli , which only share 85% sequence identity, were shown to display up to 23% of introgression in their core genome [ 18 ] for a single pair of strains. Our estimates revealed that 29% of the core genome of C. jejuni contains introgressed sequences from C. coli (cumulatively across all strains), which is in close agreement with the previous estimate. The Neisseria genus represents another interesting case of porous species boundaries. The species of this genus were previously reported to engage extensively in genetic exchange and this had been attributed to the presence of numerous Neisseria species living in the same ecological niches in the human body [ 16 , 38 , 39 , 40 ]. In agreement with previous studies, our results show that N. meningitidis and N. lactamica— which both colonize the human nasopharynx—share higher levels of introgressed DNA in their core genes relative to non-pathogenic Neisseria species. In contrast, we inferred that 32% of the core genome of N. meningitidis contains introgressed sequences from its closest relative N. gonorrhoeae although these two species do not typically cohabit the same ecological niche [ 40 , 41 ]. These findings indicate that many species appear to engage in introgression in a manner that is not always in agreement with their known ecology, providing new insights into bacterial evolution and speciation [ 18 , 42 , 43 ]. Overall, our results indicate that introgression is a pervasive process shaping the evolution of the core genome of bacterial species.
The amount of transferred DNA between species’ core genomes was found to vary extensively based on sequence divergence (Fig. 4 a). A minority of species’ pairs showed little to no signs of introgression despite sharing high sequence identity, e.g., >90% (Fig. 4 a). In contrast, most species display a positive exponential relationship between sequence relatedness and levels of introgression (Fig. 4 a). The same trend was observed when using different identity thresholds to define introgressed fragments (Additional file 2 : Fig. S8). Note that our method was designed to avoid inferring identical fragments as introgression when they actually result from vertical evolution, and only strains that are more closely related to one another rather than to the candidate species were considered for this analysis. The amount of vertically inherited fragments falsely inferred as introgression is expected to be negligible, but the exact number depends on the levels of divergence between species and on the levels of polymorphisms of each species. For simplicity, the number of identical 100bp segments found between simulated genome pairs was generated to represent the maximal theoretical number of DNA segments that could result from vertical evolution (Fig. 4 c, dashed blue line). We observed that most species pairs present higher levels of identical DNA segments than expected based on our upper limit of vertically inherited segments (Fig. 4 c). Findings indicate that, despite a clear reduction in gene flow, many species still engage in some levels of DNA exchange in their core genomes and that the frequency of introgression is intimately related to sequence divergence.

Introgression between species and MEPS. a Introgression scores ( S i ) between candidate and reference species that were not reclassified as part of the same species. S i represents the fraction of the core genome that shows evidence of introgression between the candidate species and at least one genome of the reference species. Introgressed regions were defined as 100bp fragments that were more similar to the candidate species than at least one of the genomes of the reference species. Only introgressed fragments with ≥95% sequence identity between the candidate and reference species were considered as introgressed. Coefficient correlation ρ and P -value were estimated with Spearman’s rank correlation test. b Simulated frequency of identical Minimal Efficient Processing Segments (MEPS) expected relative to sequence identity. Different lines represent MEPS ranging from 20 to 90bp. c Introgression scores ( S i ) computed for recent introgression events. S i was computed as described above between candidate and reference species that were not reclassified as part of the same species. Only introgressed fragments presenting 100% sequence identity between the candidate and reference species were considered as introgressed. The red line corresponds to a MEPS size of 40bp and the dashed blue line represents the expected number of 100bp identical fragments for a pair of genomes
A model of gene flow shaping species boundaries
Several studies have experimentally quantified recombination rate relative to sequence divergence in Escherichia , Bacillus and Streptococcus and a similar exponential relationship was observed [ 44 , 45 , 46 , 47 ]. One proposed hypothesis [ 48 ] was based on the observation that homologous recombination requires the presence of nearly identical DNA fragments called MEPS (minimal efficient processing segment) to initiate recombination [ 49 ]. The size of the MEPS has been experimentally shown to vary between 25 and 90bp across species [ 49 ]. We simulated multiple sets of genomes with different levels of divergence and identified the frequency of different-sized MEPS (Fig. 4 b). We found that the observed relationship between introgression and sequence divergence could be explained by the availability of MEPS. A MEPS size of 40bp likely represents the average size required to initiate recombination across species (Fig. 4 c, red line), although MEPS size likely varies across lineages. A simple model might account for the overall patterns of introgression observed across bacteria: as genomes diverge, the density of MEPS available to initiate recombination decreases. Due to the exponential shape of this relationship, gene flow appears to be sharply reduced when genomes reach 2–10% of sequence divergence. This level of genome divergence approximately corresponds to the threshold of ~95% genome sequence identity that is commonly used to define bacterial species [ 4 , 50 , 51 ]. This finding may explain why 95% ANI is commonly judged an adequate threshold to define species boundaries as a sequence identity of roughly 95% effectively interrupts gene flow and could therefore disrupt the genetic cohesiveness in most species. However, the exact sequence identity that confers an effective interruption of gene flow appears to vary across species (Fig. 2 b, c), typically ranging from 90 to 98% sequence identity and this is likely due to various MEPS sizes required to initiate homologous recombination across species. These results further support some key aspects of the fragmented speciation model [ 52 ], where different regions of the genomes may undergo independent genetic isolation due to sequence divergence and ecological adaptation, which would eventually lead to the interruption of gene flow.
Biological bacterial species can be defined based on the signal of gene flow, and we argue that this constitutes a theoretical framework upon which a biologically-relevant species concept of bacteria can be built. Defining bacterial species is key to many studies such as those focusing on population genetics, ecology, and pan-genome evolution. Although some bacterial species appear to be truly asexual, those are likely as rare and as short-lived as in multicellular eukaryotes [ 27 ]. A ~95% genome sequence identity threshold is commonly used as an approximation to define bacterial species and our results indicate that this threshold appears to vary from 90 to 98% sequence identity across lineages when defining species based on gene flow. Our results may therefore provide an explanation for why the ~95% genome sequence identity threshold has been judged a good approximation in many studies to define species based on empirical observation [ 4 , 50 , 51 ]. Previous studies reported an exponential relationship between homologous recombination and sequence divergence in several bacterial species using experimental settings [ 44 , 45 , 46 , 47 ]. Our results indicate that the availability of MEPS is likely a universal feature shaping gene flow and speciation in bacteria. Many works have emphasized the role of ecology and selection in bacterial speciation [ 42 , 43 ] and our findings do not challenge these results. Although niche specialization, physical barriers, and selection might be the primary causes of speciation in bacteria, our study suggests that barriers of gene flow might only become effective and irreversible once genomes have reached a certain level of divergence.
All analyzed genomes were downloaded from the GenBank database ftp.ncbi.nlm.nih.gov/genomes / (September 2018). All named species—as named on GenBank—with ≥15 completely assembled genomes were downloaded. This dataset initially included 84,078 bacterial and archaeal genomes from 331 named species according to species designations on the NCBI website (Additional file 1 : Table S1). Protein-coding genes of each genome were extracted based on the annotations. From the original set of 331 named species, only those represented by at least 15 genomes remaining after filtering for missing or incomplete annotations were conserved (Additional file 1 : Table S1). Six species presented very large genomic data (>2,500 genomes), i.e., Acinetobacter baumannii , Escherichia coli , Klebsiella pneumoniae , Salmonella enterica , Staphylococcus aureus , and Streptococcus pneumoniae , and 500 genomes were therefore randomly selected for each of these named species in order to reduce the computational load (Additional file 1 : Table S1). This total database resulted in a total of 30,694 genomes from 247 named species with ≥15 genomes for each species represents our dataset of reference species (Additional file 5 : Dataset S1), against which related species— candidate species —have been compared. Note that our overall dataset contained a single archaeal species and we therefore referred to our dataset as “bacteria” instead of “prokaryotes” to avoid generalization to all prokaryotes since our dataset includes a single archaeal species. We then downloaded one fully assembled genome sequence and the corresponding annotations for 2595 candidate species (Additional file 6 : Dataset S2). Candidate species were selected by choosing species within the same genus of the reference species (e.g., since Bacillus cereus was present in our list of reference species, all other Bacillus species were used as candidate species). Finally, because bacterial classification can be inconsistent, we also identified genera sharing high sequence identity with one another using a set of 44 universally conserved proteins as in [ 53 ] and pairwise distances were computed using RAxML v8 with the PROTGAMMAAUTO option [ 54 ]. Genera sharing ≤5% protein sequence distances were considered as potentially misclassified genera. For each reference species of these genera, all the species from the related genera were also used as candidate species. Two groups of genera were found highly related: (i) Citrobacter , Enterobacter , Escherichia , Klebsiella , Salmonella , and Shigella and (ii) Mycobacterium and Mycobacteroides .
Definition of core genomes of the reference species
For each reference species, the core genome was built using CoreCruncher as previously described [ 55 ] with Usearch Global v8.0 [ 56 ] and the stringent option. CoreCruncher was used because it is fast and because it includes a test to exclude potential paralogs and xenologs from the core genome. Orthologs were defined with >70% protein sequence identity and >80% sequence length conservation and all other parameters were set to default. The core genome was defined as the set of single-copy orthologs found in at least 85% of the genomes within each species. Protein sequences of each core gene were then aligned using Muscle v3.8.31 [ 57 ] with default parameters. Because Muscle was unable to align large sequence files, Mafft v7.407 [ 58 ] was used for the species containing ≥1000 genomes. Protein alignments were then reverse-translated into their corresponding nucleotide sequences. Finally, the nucleotide alignments of all the core genes of each named species were concatenated into a single large alignment as previously described [ 59 ].
Definition of species based on ANI of core genes and gene flow
The core genome concatenates of each of the 247 reference species were used to estimate the ANI of core genes for all genome pairs. This method differs from more traditional ANI, e.g., using FastANI [ 4 ], in the fact that traditional ANI represents the average nucleotide identity of all orthologous genes shared by any two genomes while the ANI of core genes represents the average nucleotide identity of the core genes shared by a pair of genomes. The methods are very similar in concept, but the ANI of core genes is a slightly more stringent metric as core genes are usually evolving slower than accessory genes [ 19 ]. Pairwise ANI of core gene values were computed using the distmat tool of EMBOSS version 6.6.0.0 [ 60 ], which calculates the pairwise nucleotide identities from the alignment as previously described [ 19 ]. Then, single linkage clustering was performed as described in [ 19 ]: all genome pairs with an ANI of core gene similarity cutoff of 94% or higher were joined together and clustered into de novo species (ANI species) (Additional file 1 : Table S1).
Gene flow was then inferred using the distance-based method implemented in ConSpeciFix (8). Briefly, the matrix of pairwise distances D was built using RAxML v8 [ 54 ] with the GTR + GAMMA model for each core genome concatenate. This matrix of distances was used to infer homoplasic alleles ( h ) and non-homoplasic alleles ( m ) as described in (8). A genome resampling analysis was conducted for each dataset in order to identify the presence of potential outliers, i.e., genomes that do not engage in gene flow with the rest of the population. Groups of genomes were considered part of the same biological species when found to engage in gene flow, whereas genomes whose inclusion led to a substantial and significant drop in gene flow ( h/m ) based on the exclusion criterion were excluded from the biological species as previously described [ 9 , 30 ]. First, gene flow was estimated using the dataset of reference species. From each of these named species, genomes that led to a substantial drop in gene flow were identified by a significant and substantial decline in h/m ratios (Wilcoxon test, P < 0.0001). These genomes were then removed from the dataset, and all remaining genomes were considered members of the same BSC-defined species.
Our final classification of reference species was based on the results of both methods—ANI of core genes and gene flow delimitation. Genomes that were excluded by at least one of these methods were excluded from the reference species. The final dataset of redefined reference species was composed of more than 30,000 genomes across 227 species (Additional file 5 : Dataset S1, Additional file 1 : Table S1). This dataset is referred as redefined BSC species (but the ANI of core genes was also used to redefine species boundaries). In addition, when named species regrouped more than one cluster with ≥15 genomes, the cluster containing the reference genome on NCBI was selected to represent the reference species and one genome of the rest of the other clusters in the same named species was added to the dataset of candidate species in case it would be reclassified as a part of a different reference species.
Inference of clonal species
Simulation approach.
For each species, simulations were conducted in the absence of recombination to estimate the expected number of homoplasies introduced by convergent mutations. The goal of this analysis was to determine which species present h/m ratios compatible with a purely clonal model of evolution. First, a maximum likelihood phylogenetic tree for each of the 227 species was built using the total core genome alignment of each species using RAxML v8 [ 54 ] with a GTR + Gamma model. Several summary statistics were extracted from the alignment and from the tree for each species: GC-content, core genome alignment length, average pairwise nucleotide diversity ( π ), levels of polymorphisms across codon positions and the transition/transversion ratio ( κ ). We then used CoreSimul [ 61 ] to generate forward-in-time simulations of the core genome of each species using the parameters specific to each species. Simulations were initiated by generating a random core genome using the length and the GC content estimated for each species. The topology and the branch lengths of the tree were used to simulate core genome evolution without recombination (recombination rate ρ was set to zero) under a K2P and codon model using species-specific parameters: the κ parameter estimated for each species and the relative levels of polymorphisms estimated across the three codon positions were used to simulate substitutions with specific rates across the three codon positions specific to each species. Phylogenetic programs infer homoplasic alleles as independent mutations, although they often result from recombination events [ 61 ]. Therefore, the branch lengths of the trees systematically overestimate the amount of polymorphisms present in the genomes when recombination is present [ 61 , 62 ]. To address this issue, the genome simulations of each species were conducted multiple times ( n =99) with different rescaling coefficients as explained in [ 62 ]. We then estimated the pairwise ANI of core genes for each simulation replicate and these values were compared to the pairwise ANI of the real core genome of the corresponding species. For each species, the simulated replicate presenting the most similar nucleotide diversity to the real dataset was then selected as the most realistic simulation. Finally, this simulated set of genomes was used to compute the h/m ratio for each species. This h/m ratio then was used to infer the amount of homoplasies expected under clonal evolution specific to each species. The real h/m ratios were compared to the h/m ratios of the datasets simulated without recombination to infer which species were clonal (Additional file 2 : Fig. S1a, Additional file 3 : Table S2). Assuming that all the species in our dataset were strictly clonal, we would expect the real and simulated ratios to be very similar ( y=x ). Most species presented clearly different h/m ratios between real and simulated data (Additional file 2 : Fig. S1a, Additional file 3 : Table S2). Using a resampling analysis, the real h/m ratio was re-computed 100 times for each species by excluding exactly one genome for each resampling. The standard deviation of the h/m ratios (SD) calculated with the resampling analysis—based on the standard deviation—was used to define the limit between clonal species and non-clonal species (Additional file 2 : Fig. S1a, Additional file 3 : Table S2). Very similar numbers of species were defined as clonal when using a threshold of 2.SD, 3.SD or 4.SD. The more conservative threshold of 3.SD was then selected to define clonal species with this approach.
Linkage disequilibrium approach
Linkage disequilibrium (LD) analysis was conducted in the core concatenate of each redefined reference species as for the simulation study. We used the presence or absence of LD signal to infer the clonality for each of the 227 species. LD was measured using r 2 between all pairs of biallelic loci A/a and B/b as: r 2 = \(\frac{{\left( pAB- pA. pB\right)}^2}{pA.\left(1- pA\right). pB.\left(1- pB\right)}\) with p AB the proportion of haplotypes AB, p A the proportion of alleles A, p B the proportion of alleles B and such as p a =1-p A and p b =1-p B . Positions in the alignments with ≥25% missing sites due to indels were not included. Because singletons can lead to substantially underestimate the signal of recombination, biallelic sites were only included in the analysis when the least frequent allele was found in at least two genomes. LD was estimated in the core genome concatenate of each redefined reference species using a scanning window of 1,000bp. We considered that a significant decrease in r 2 relative to genomic distance between alleles was indicative of the presence of gene flow, whereas the absence of correlation between r 2 and genomic distances was indicative of the absence of gene flow. Correlations were assessed with Spearman’s ρ and a conservative p -value threshold of P <0.0001—which accounts for multiple testing ( α =0.0226)—was defined to infer significant correlations (Additional file 2 : Fig. S1b).
Inference of synonymous and non-synonymous alleles
To test for the potential impact of adaptive evolution on the prevalence of homoplasies, we classified homoplasic alleles ( h ) and non-homoplasic alleles ( m ) as synonymous or non-synonymous across the core genome of all reference species. The core genome of each reference species is a concatenate of genes that were reverse transcribed from protein alignments (see above) and our inference of homoplasies further provide the position of these alleles along the concatenate. To avoid ambiguities, we focused our analysis on codons with a single polymorphic site. The analysis was further restricted to codons whose polymorphic site was bi-allelic, also to avoid ambiguous inferences. We then computed the number of homoplasic alleles and non-homoplasic alleles found at synonymous and non-synonymous positions, respectively. Because reference species vary in core genome size and levels of polymorphisms, we reported the results for the species where ≥100 homoplasic alleles and ≥100 non-homoplasic alleles could be analyzed.
Definition of the core genome of the reference and candidate species
We used the ConSpeciFix pipeline [ 30 ] to build the core genome shared between each reference species and each related candidate species (i.e., the species belonging to the same genus or to a closely related genus, see above) using a single randomly selected genome for the candidate species. Briefly, ConSpeciFix compares each core gene of the reference species to the genome of the candidate species using Usearch Global v8.0 [ 56 ] with ≥70% protein identity and ≥80% sequence length conservation. Orthologs were defined as best bidirectional hits and considered part of the shared core genome if found as single-copy as in [ 9 ]. Protein sequences of each core genes were aligned and reverse-translated into nucleotides as described above. The shared core genome of each pair of reference + candidate species was then concatenated into a single large alignment. This step resulted in a total of 13,209 core genome concatenates (Additional file 4 : Table S3), each corresponding to a specific pair of reference + candidate species. We also calculated the ANI of core genes for all pairs of genomes between reference and candidate species using the same approach as described above.
Detection of gene flow between species
We tested for the presence of gene flow between each of the BSC-defined species and each candidate species. For each comparison of a candidate species against a BSC-defined reference species, the core genome concatenate for the reference + candidate species was used to infer gene flow using ConSpeciFix [ 30 ] as described above. The core genome concatenate was used to compute a distance matrix using RAxML version 8.2.12 (10). From these distances, the ratio of homoplasic to non-homoplasic alleles ( h/m ) was computed for i) the BSC-defined reference species alone and ii) the BSC-defined reference species + the candidate genome. Subsampling analyses were conducted as above. From this step, graphs and statistics comparing h/m ratios between the genomes of each BSC-defined reference species with and without the candidate genome were inferred as previous described [ 30 ]. The candidate species was inferred as a distinct BSC species when a significant and substantial reduction of gene flow was detected based on h/m ratios (Wilcoxon test, P <0.0001). When no clear reduction of gene flow was observed, the reference species and the candidate species were considered as putatively part of the same biological species and further tested for convergent mutations (see below).
Convergent mutation test
Because our procedure is comparing various genomes, some comparisons can occasionally involve species with substantial genomic divergence. As genomes accumulate mutations during divergence, the frequency of convergent mutations increases, and this leads to the accumulation of homoplasic alleles that are the result of mutations rather than gene flow. To control for this, we simulated genome sequences for each dataset of reference + candidate species. The goal of this analysis is to generate a simulated genome sequence with similar sequence divergence and characteristics as the genome of the candidate species relative to the reference species and to estimate the ratio h/m 0 expected to result from convergent mutations alone (Additional file 2 : Fig. S3). Each sequence was evolved in silico with mutations but without gene flow . This simulated sequence was then used to estimate the ratio h/m 0 against the reference species. The estimated values of h/m 0 are then compared to the real h/m values obtained between the candidate species and the reference species ( h/m cand ). We considered cases where h/m 0 is similar to h/m cand as indicative that the signal of gene flow is actually driven by convergent mutations rather than gene flow.
First, the consensus sequence of the core genome concatenate of the BSC-defined species is generated by selecting the most frequent allele at each site. Random point mutations are then introduced in silico with a Jukes and Cantor model until the same sequence divergence is obtained as the one observed between the genomes of the BSC-defined species and the candidate genome. This step was conducted for each of the comparisons of BSC-redefined reference species against candidate species (13,437 comparisons). The resulting concatenate was then analyzed with the ConSpeciFix process as described above to infer h/m 0 ratios. The candidate species was then considered as truly engaging in gene flow with the reference species when h/m cand was found significantly higher than h/m 0 (Wilcoxon test, P <0.0001).
From these metrics, we also derived the metric h/m norm , which quantifies gene flow between the candidate and the reference species rescaled by the amount of gene flow observed in the reference species alone ( h/m ref ), i.e., without the candidate species and by the expected amount of homoplasies introduced by convergent mutations, or, against the sequence simulated without gene flow. We expressed h/m norm = ( h/m cand − h/m 0 ) / ( h/m ref − h/m 0 ) so that h/m norm = 0 corresponds to h/m cand = h/m 0 and h/m norm = 1 corresponds to h/m cand = h/m ref .
Introgression analysis
Introgression was defined as fragments of DNA exchanged by gene flow between related species. Introgression was inferred using the concatenate of the shared core genome between each BSC-defined reference species and each candidate species (13,437 concatenates analyzed). We used a non-overlapping sliding window of 100bp to estimate the identity score of the shared core genome concatenate. For each window, the average, the minimal, and the maximal nucleotide identity were calculated for (i) the genomes of the redefined reference species alone and (ii) between the genome of the candidate species and the genomes of the reference species. We considered that a 100-bp fragment was introgressed when at least one genome of the reference species was more similar to the candidate genome than one of the other genomes of the reference species. The introgression score S i was then defined as the fraction of the core genome that has been found introgressed between the candidate species and at least one genome of the reference species. Introgression scores were also computed by imposing different thresholds of sequence identity between the candidate species and the reference species for a fragment to be considered introgressed: 90%, 95%, 98%, and 100% (Additional file 2 : Fig. S7). These different thresholds represent increasingly ancient introgression events. Importantly, this analysis is based on the assumption that the difference genomes of the reference species are more closely related to one another relative to the genome of the candidate species. Therefore, we did not compute the introgression score when one or more genomes of the reference species were found more related to the candidate species than to another genome of the reference species. The matrix of maximum likelihood distances ( D) computed by RAxML was used to infer genome distances for each pair of candidate/reference species (see above).
MEPS simulations
Pairs of 1Mb sequences with various levels of sequence identity (from 80 to 100%) were simulated with a Jukes and Cantor model of substitution, no indels and a GC content of 50%. For each pair of sequences, the number of potential MEPS was defined as the number of strictly identical segments of DNA shared between the two sequences. Identical fragments were identified using a scanning window of size 20bp, 30bp, 40bp, 50bp, 60bp, 70bp, 80bp, 90bp, and 100bp.
Availability of data and materials
Genomes used in this study are listed in Dataset S1 (Additional file 5 ) and Dataset S2 (Additional file 6 ) and are freely available on GenBank at https://www.ncbi.nlm.nih.gov/genome/ . All the core genome datasets used in this study are available at Kaggle [ 63 , 64 , 65 , 66 , 67 ].
Fraser C, Alm EJ, Polz MF, Spratt BG, Hanage WP. The bacterial species challenge: making sense of genetic and ecological diversity. Science. 2009;323:741–6. https://doi.org/10.1126/science.1159388 .
Article CAS PubMed Google Scholar
Doolittle WF. Population genomics: how bacterial species form and why they don’t exist. Curr Biol. 2012;22:R451–3. https://doi.org/10.1016/j.cub.2012.04.034 .
Konstantinidis KT, Tiedje JM. Genomic insights that advance the species definition for prokaryotes. Proc Natl Acad Sci U S A. 2005;102:2567–72. https://doi.org/10.1073/pnas.0409727102 .
Article CAS PubMed PubMed Central Google Scholar
Jain C, Rodriguez-R LM, Phillippy AM, Konstantinidis KT, Aluru S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat Commun. 2018;9:5114. https://doi.org/10.1038/s41467-018-07641-9 .
Smith JM, Smith NH, O’Rourke M, Spratt BG. How clonal are bacteria? Proc Natl Acad Sci. 1993;90:4384–8. https://doi.org/10.1073/pnas.90.10.4384 .
Ochman H, Lerat E, Daubin V. Examining bacterial species under the specter of gene transfer and exchange. Proc Natl Acad Sci U S A. 2005;102(Suppl 1):6595–9. https://doi.org/10.1073/pnas.0502035102 .
Cadillo-Quiroz H, Didelot X, Held NL, Herrera A, Darling A, Reno ML, et al. Patterns of Gene Flow Define Species of Thermophilic Archaea. Barton NH, editor. PLoS Biol. 2012;10:e1001265. https://doi.org/10.1371/journal.pbio.1001265 .
Shapiro BJ, Friedman J, Cordero OX, Preheim SP, Timberlake SC, Szabó G, et al. Population Genomics of Early Events in the Ecological Differentiation of Bacteria. Science. 2012;336:48–51. https://doi.org/10.1126/science.1218198 .
Bobay L-M, Ochman H. Biological Species Are Universal across Life’s Domains. Genome Biol Evol. 2017;9:491–501. https://doi.org/10.1093/gbe/evx026 .
Article PubMed Central Google Scholar
Arevalo P, VanInsberghe D, Elsherbini J, Gore J, Polz MF. A Reverse Ecology Approach Based on a Biological Definition of Microbial Populations. Cell. 2019;178:820–834.e14. https://doi.org/10.1016/j.cell.2019.06.033 .
Olm MR, Crits-Christoph A, Diamond S, Lavy A, Matheus Carnevali PB, Banfield JF. Consistent Metagenome-Derived Metrics Verify and Delineate Bacterial Species Boundaries. mSystems. 2020;5:e00731–19. https://doi.org/10.1128/mSystems.00731-19 .
Polz MF, Alm EJ, Hanage WP. Horizontal gene transfer and the evolution of bacterial and archaeal population structure. Trends Genet TIG. 2013;29:170–5. https://doi.org/10.1016/j.tig.2012.12.006 .
VanInsberghe D, Arevalo P, Chien D, Polz MF. How can microbial population genomics inform community ecology? Philos Trans R Soc B Biol Sci. 2020;375:20190253. https://doi.org/10.1098/rstb.2019.0253 .
Article Google Scholar
Bobay L-M. The Prokaryotic Species Concept and Challenges. In: Tettelin H, Medini D, editors. Pangenome Divers Dyn Evol Genomes. Cham: Springer International Publishing; 2020. p. 21–49. https://doi.org/10.1007/978-3-030-38281-0_2 .
Chapter Google Scholar
Hedge J, Wilson DJ. Bacterial phylogenetic reconstruction from whole genomes is robust to recombination but demographic inference is not. mBio. 2014;5:e02158. https://doi.org/10.1128/mBio.02158-14 .
Article PubMed PubMed Central Google Scholar
Hanage WP, Fraser C, Spratt BG. Fuzzy species among recombinogenic bacteria. BMC Biol. 2005;3:6. https://doi.org/10.1186/1741-7007-3-6 .
Sheppard SK, McCarthy ND, Falush D, Maiden MCJ. Convergence of Campylobacter Species: Implications for Bacterial Evolution. 2008;320:4. https://doi.org/10.1126/science.1155532 .
Sheppard SK, Didelot X, Jolley KA, Darling AE, Pascoe B, Meric G, et al. Progressive genome-wide introgression in agricultural Campylobacter coli . Mol Ecol. 2013;22:1051–64. https://doi.org/10.1111/mec.12162 .
Wittouck S, Wuyts S, Meehan CJ, van Noort V, Lebeer S. A Genome-Based Species Taxonomy of the Lactobacillus Genus Complex. mSystems. 2019;4:e00264–19. https://doi.org/10.1128/mSystems.00264-19 .
Seth-Smith HMB, Busó LS, Livingstone M, Sait M, Harris SR, Aitchison KD, et al. European Chlamydia abortus livestock isolate genomes reveal unusual stability and limited diversity, reflected in geographical signatures. BMC Genomics. 2017;18:344. https://doi.org/10.1186/s12864-017-3657-y .
Holzapfel M, Girault G, Keriel A, Ponsart C, O’Callaghan D, Mick V. Comparative Genomics and in vitro Infection of Field Clonal Isolates of Brucella melitensis Biovar 3 Did Not Identify Signature of Host Adaptation. Front Microbiol. 2018;9:2505. https://doi.org/10.3389/fmicb.2018.02505 .
Gogol EB, Cummings CA, Burns RC, Relman DA. Phase variation and microevolution at homopolymeric tracts in Bordetella pertussis. BMC Genomics. 2007;8:122. https://doi.org/10.1186/1471-2164-8-122 .
Toprak E, Veres A, Michel J-B, Chait R, Hartl DL, Kishony R. Evolutionary paths to antibiotic resistance under dynamically sustained drug selection. Nat Genet. 2011;44:101–5. https://doi.org/10.1038/ng.1034 .
Barroso-Batista J, Sousa A, Lourenço M, Bergman M-L, Sobral D, Demengeot J, et al. The First Steps of Adaptation of Escherichia coli to the Gut Are Dominated by Soft Sweeps. PLoS Genet. 2014;10:e1004182. https://doi.org/10.1371/journal.pgen.1004182 .
Ramiro RS, Durão P, Bank C, Gordo I. Low mutational load and high mutation rate variation in gut commensal bacteria. PLoS Biol. 2020;18:e3000617. https://doi.org/10.1371/journal.pbio.3000617 .
Hershberg R, Petrov DA. Selection on codon bias. Annu Rev Genet. 2008;42:287–99. https://doi.org/10.1146/annurev.genet.42.110807.091442 .
Maynard SJ. The Evolution of Sex. Cambridge: Cambridge University Press; 1978.
Google Scholar
Bennett GM, Moran NA. Heritable symbiosis: The advantages and perils of an evolutionary rabbit hole. Proc Natl Acad Sci U S A. 2015;112:10169–76. https://doi.org/10.1073/pnas.1421388112 .
Chong RA, Moran NA. Evolutionary loss and replacement of Buchnera, the obligate endosymbiont of aphids. ISME J. 2018;12:898–908. https://doi.org/10.1038/s41396-017-0024-6 .
Bobay L-M, Ellis BS-H, Ochman H. ConSpeciFix: classifying prokaryotic species based on gene flow. Hancock J, editor. Bioinformatics. 2018;34:3738–40. https://doi.org/10.1093/bioinformatics/bty400 .
Yahara K, Didelot X, Jolley KA, Kobayashi I, Maiden MCJ, Sheppard SK, et al. The Landscape of Realized Homologous Recombination in Pathogenic Bacteria. Mol Biol Evol. 2016;33:456–71. https://doi.org/10.1093/molbev/msv237 .
Didelot X, Méric G, Falush D, Darling AE. Impact of homologous and non-homologous recombination in the genomic evolution of Escherichia coli. BMC Genomics. 2012;13:256. https://doi.org/10.1186/1471-2164-13-256 .
Chen L, Cai Y, Zhou G, Shi X, Su J, Chen G, et al. Rapid Sanger sequencing of the 16S rRNA gene for identification of some common pathogens. PLoS One. 2014;9:e88886. https://doi.org/10.1371/journal.pone.0088886 .
Pettengill EA, Pettengill JB, Binet R. Phylogenetic Analyses of Shigella and Enteroinvasive Escherichia coli for the Identification of Molecular Epidemiological Markers: Whole-Genome Comparative Analysis Does Not Support Distinct Genera Designation. Front Microbiol. 2016;6:1573. https://doi.org/10.3389/fmicb.2015.01573 .
Maderankova D, Jugas R, Sedlar K, Vitek M, Skutkova H. Rapid Bacterial Species Delineation Based on Parameters Derived From Genome Numerical Representations. Comput Struct Biotechnol J. 2019;17:118–26. https://doi.org/10.1016/j.csbj.2018.12.006 .
Mahenthiralingam E, Baldwin A, Dowson CG. Burkholderia cepacia complex bacteria: opportunistic pathogens with important natural biology. J Appl Microbiol. 2008;104:1539–51. https://doi.org/10.1111/j.1365-2672.2007.03706.x .
Mannaa M, Park I, Seo Y-S. Genomic Features and Insights into the Taxonomy, Virulence, and Benevolence of Plant-Associated Burkholderia Species. Int J Mol Sci. 2018;20:E121. https://doi.org/10.3390/ijms20010121 .
Linz B, Schenker M, Zhu P, Achtman M. Frequent interspecific genetic exchange between commensal Neisseriae and Neisseria meningitidis. Mol Microbiol. 2000;36:1049–58. https://doi.org/10.1046/j.1365-2958.2000.01932.x .
Marri PR, Paniscus M, Weyand NJ, Rendón MA, Calton CM, Hernández DR, et al. Genome sequencing reveals widespread virulence gene exchange among human Neisseria species. PLoS One. 2010;5:e11835. https://doi.org/10.1371/journal.pone.0011835 .
Hanage WP. Fuzzy species revisited. BMC Biol. 2013;11:41. https://doi.org/10.1186/1741-7007-11-41 .
Quillin SJ, Seifert HS. Neisseria gonorrhoeae host adaptation and pathogenesis. Nat Rev Microbiol. 2018;16:226–40. https://doi.org/10.1038/nrmicro.2017.169 .
Caro-Quintero A, Rodriguez-Castaño GP, Konstantinidis KT. Genomic insights into the convergence and pathogenicity factors of Campylobacter jejuni and Campylobacter coli species. J Bacteriol. 2009;191:5824–31. https://doi.org/10.1128/JB.00519-09 .
Lefébure T, Bitar PDP, Suzuki H, Stanhope MJ. Evolutionary dynamics of complete Campylobacter pan-genomes and the bacterial species concept. Genome Biol Evol. 2010;2:646–55. https://doi.org/10.1093/gbe/evq048 .
Majewski J, Cohan FM. The Effect of Mismatch Repair and Heteroduplex Formation on Sexual Isolation in Bacillus. Genetics. 1998;148:13–8. https://doi.org/10.1093/genetics/148.1.13 .
Majewski J, Cohan FM. Adapt Globally, Act Locally: The Effect of Selective Sweeps on Bacterial Sequence Diversity. Genetics. 1999;152:1459–74. https://doi.org/10.1093/genetics/152.4.1459 .
Majewski J, Cohan FM. DNA Sequence Similarity Requirements for Interspecific Recombination in Bacillus. Genetics. 1999;153:1525–33. https://doi.org/10.1093/genetics/153.4.1525 .
Majewski J, Zawadzki P, Pickerill P, Cohan FM, Dowson CG. Barriers to Genetic Exchange between Bacterial Species: Streptococcus pneumoniae Transformation. J Bacteriol. 2000;182:1016–23. https://doi.org/10.1128/JB.182.4.1016-1023.2000 .
Majewski J. Sexual isolation in bacteria. FEMS Microbiol Lett. 2001;199:161–9. https://doi.org/10.1111/j.1574-6968.2001.tb10668.x .
Shen P, Huang HV. Homologous recombination in Escherichia coli: dependence on substrate length and homology. Genetics. 1986;112:441–57. https://doi.org/10.1093/genetics/112.3.441 .
Murray CS, Gao Y, Wu M. Re-evaluating the evidence for a universal genetic boundary among microbial species. Nat Commun. 2021;12:4059. https://doi.org/10.1038/s41467-021-24128-2 .
Rodriguez-R LM, Jain C, Conrad RE, Aluru S, Konstantinidis KT. Reply to: “Re-evaluating the evidence for a universal genetic boundary among microbial species.”. Nat Commun. 2021;12:4060. https://doi.org/10.1038/s41467-021-24129-1 .
Retchless AC, Lawrence JG. Phylogenetic incongruence arising from fragmented speciation in enteric bacteria. Proc Natl Acad Sci U S A. 2010;107:11453–8. https://doi.org/10.1073/pnas.1001291107 .
Bobay LM, Ochman H. Factors driving effective population size and pan-genome evolution in bacteria. BMC Evol Biol. 2018;18:153. https://doi.org/10.1186/s12862-018-1272-4 .
Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–3. https://doi.org/10.1093/bioinformatics/btu033 .
Harris CD, Torrance EL, Raymann K, Bobay L-M. CoreCruncher: fast and robust construction of core genomes in large prokaryotic datasets. Ouangraoua A, editor. Mol Biol Evol. 2020:msaa224. https://doi.org/10.1093/molbev/msaa224 .
Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–1. https://doi.org/10.1093/bioinformatics/btq461 .
Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–7. https://doi.org/10.1093/nar/gkh340 .
Katoh K, Standley DM. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol Biol Evol. 2013;30:772–80. https://doi.org/10.1093/molbev/mst010 .
Bobay L-M, Ochman H. Impact of Recombination on the Base Composition of Bacteria and Archaea. Mol Biol Evol. 2017;34:2627–36. https://doi.org/10.1093/molbev/msx189 .
Rice P, Longden I, Bleasby A. EMBOSS: The European molecular biology open software suite. Trends Genet TIG. 2000;16:276–7. https://doi.org/10.1016/s0168-9525(00)02024-2 .
Didelot X, Wilson DJ. ClonalFrameML: Efficient Inference of Recombination in Whole Bacterial Genomes. Prlic A, editor. PLoS Comput Biol. 2015;11:e1004041. https://doi.org/10.1371/journal.pcbi.1004041 .
Bobay L-M. CoreSimul: a forward-in-time simulator of genome evolution for prokaryotes modeling homologous recombination. BMC Bioinformatics. 2020;21:264. https://doi.org/10.1186/s12859-020-03619-x .
Diop A, Torrance EL, Stott CM, Bobay LM. Gene Flow and Introgression are Pervasive Forces Shaping the Evolution of Bacterial Species. Diop-genome-biology-2022. Kaggle; 2022. https://doi.org/10.34740/KAGGLE/DS/2546741 .
Book Google Scholar
Diop A, Torrance EL, Stott CM, Bobay LM. Gene Flow and Introgression are Pervasive Forces Shaping the Evolution of Bacterial Species. Diop-genome-biology-2022-2. Kaggle; 2022. https://doi.org/10.34740/KAGGLE/DSV/4341662 .
Diop A, Torrance EL, Stott CM, Bobay LM. Gene Flow and Introgression are Pervasive Forces Shaping the Evolution of Bacterial Species. Diop-genome-biology-2022-3. Kaggle; 2022. https://doi.org/10.34740/KAGGLE/DSV/4342322 .
Diop A, Torrance EL, Stott CM, Bobay LM. Gene Flow and Introgression are Pervasive Forces Shaping the Evolution of Bacterial Species. Diop-genome-biology-2022-4. Kaggle; 2022. https://doi.org/10.34740/KAGGLE/DSV/4342067 .
Diop A, Torrance EL, Stott CM, Bobay LM. Gene Flow and Introgression are Pervasive Forces Shaping the Evolution of Bacterial Species. Diop-genome-biology-2022-5. Kaggle; 2022. https://doi.org/10.34740/KAGGLE/DSV/4346891 .
Download references
Acknowledgements
We thank Kasie Raymann and Joseph Santin for providing feedback on the manuscript.
Review history
The review history is available as Additional file 7 .
Peer review information
Tim Sands was the primary editor of this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
This study is supported by the National Science Foundation NSF grant DEB-1831730 (LMB), the National Institutes of Health grant R01GM132137 (LMB), and DOE Computational Science Graduate Fellowship DE-SC0021110 (ELT).
Author information
Authors and affiliations.
Department of Biology, University of North Carolina Greensboro, Greensboro, North Carolina, 321 McIver Street, PO Box 26170, Greensboro, NC, 27402, USA
Awa Diop, Ellis L. Torrance, Caroline M. Stott & Louis-Marie Bobay
You can also search for this author in PubMed Google Scholar
Contributions
A.D. and LM.B. contributed to the conception and design of the project and interpretation of results. LM.B. supervised the project. A.D., LM.B., E.L.T., and C.M.S. performed analyses. A.D. and LM.B. wrote the manuscript, which was approved by all authors.
Corresponding author
Correspondence to Louis-Marie Bobay .
Ethics declarations
Ethics approval and consent to participate.
Not applicable.
Consent for publication
Competing interests.
The authors declare that they have no competing interests.
Additional information
Publisher’s note.
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: table s1..
Summary data of reference species.
Additional file 2:.
Supplementary Figures S1-S8.
Additional file 3: Table S2.
Metrics inferred for clonal species.
Additional file 4: Table S3.
Metrics inferred for the reclassification of candidate species.
Additional file 5: Dataset S1.
List of the reference species genomes.
Additional file 6: Dataset S2.
List of the candidate species genomes.
Additional file 7.
Peer review history.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/ . The Creative Commons Public Domain Dedication waiver ( http://creativecommons.org/publicdomain/zero/1.0/ ) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
Reprints and permissions
About this article
Cite this article.
Diop, A., Torrance, E.L., Stott, C.M. et al. Gene flow and introgression are pervasive forces shaping the evolution of bacterial species. Genome Biol 23 , 239 (2022). https://doi.org/10.1186/s13059-022-02809-5
Download citation
Received : 27 October 2021
Accepted : 24 October 2022
Published : 10 November 2022
DOI : https://doi.org/10.1186/s13059-022-02809-5
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
- Biological species definition
- Introgression
- Bacterial evolution
- Recombination
- Core genome
Genome Biology
ISSN: 1474-760X
- Submission enquiries: [email protected]
- General enquiries: [email protected]
If you're seeing this message, it means we're having trouble loading external resources on our website.
If you're behind a web filter, please make sure that the domains *.kastatic.org and *.kasandbox.org are unblocked.
To log in and use all the features of Khan Academy, please enable JavaScript in your browser.
Biology archive
Course: biology archive > unit 23.
- Taxonomy and the tree of life
- Species & speciation
- Biodiversity and natural selection
Genetic variation, gene flow, and new species
- Discovering the tree of life
- Understanding and building phylogenetic trees
- Phylogenetic trees
- Building a phylogenetic tree
Want to join the conversation?
- Upvote Button navigates to signup page
- Downvote Button navigates to signup page
- Flag Button navigates to signup page

Video transcript
- COVID-19 Tracker
- Biochemistry
- Anatomy & Physiology
- Microbiology
- Neuroscience
- Animal Kingdom
- NGSS High School
- Latest News
- Editors’ Picks
- Weekly Digest
- Quotes about Biology
Genetic Drift vs. Gene Flow vs. Natural Selection
Reviewed by: BD Editors
Genetic drift, gene flow, and natural selection may sound similar or even confusing to some. All three are mechanisms in the evolutionary process that have to do with alleles and/or gametes, but there are several significant differences.
Discussions about genes and natural selection usually include the term allele. An allele is just one version of a gene found at the same place (locus) on a chromosome. An example of an allele is the color of a bird’s feathers. In sexually reproducing organisms, alleles occur in pairs because the offspring receive one from each parent.
Genetic Drift
In genetic drift, alleles change frequency within a population due to random sampling. As a result, it does not produce adaptations. Two mechanisms cause genetic drift. The first is the bottle effect. This is genetic drift in a population after it has gone through a catastrophic event like a flood. The bottleneck happens when the allele frequency of a main trait in the original population is reduced because so many individuals carrying the allele have died. This causes most of the surviving population to die off, leaving a few random individuals as survivors. The other mechanism is called the founder effect. This is when a few members of a population break away and create their own group. Because of the random sampling that created the new group, the allele frequency can dramatically shift depending on the selective pressures place on the individuals.
Gene flow differs from genetic drift because it is the transfer of alleles or gametes from one population to another . It happens when a population migrates or becomes geographically isolated. This is different from the genetic drift seen with the founder effect where the new group is formed in an area that does not have an existing population .
Natural Selection
Natural selection is like genetic drift but with one major difference—it’s not random. And unlike genetic drift which can be helpful, detrimental, or have no effect, natural selection represents only positive change/adaptation. Also, natural selection is influenced by changes in environmental conditions while genetic drift is random and based on luck. One major way gene flow is different from natural selection is that gene flow helps keep alleles in a population homogenized while natural selection increases genetic variation and always moves toward creating new species.
A Quick Genetic Drift vs Gene Flow vs Natural Selection Comparison. (n.d.). In Biology Wise . Retrieved from https://biologywise.com/genetic-drift-vs-gene-flow-vs-natural-selection
Cite This Article
Subscribe to our newsletter, privacy policy, terms of service, scholarship, latest posts, white blood cell, t cell immunity, satellite cells, embryonic stem cells, popular topics, animal cell, horticulture, photosynthesis, hydrochloric acid, acetic acid, hermaphrodite.
Eighty years of gene-for-gene relationship and its applications in identification and utilization of R genes
- Review Article
- Published: 02 July 2021
- Volume 100 , article number 50 , ( 2021 )
Cite this article

- Bhavjot Kaur 1 ,
- Dharminder Bhatia ORCID: orcid.org/0000-0001-6088-3886 1 &
- G. S. Mavi 1
1683 Accesses
20 Citations
Explore all metrics
The gene-for-gene relationship of host–pathogen interaction explained by H. H. Flor in mid of the 20th century set a milestone in understanding the biochemical and genetic basis of plant diseases and several components involved in plant–pathogen interactions. It highlighted the importance of accomplishing differential sets and understanding the pathogen population structure, it further led to the identification and cloning of several resistance ( R ) genes in plants. These R genes have been deployed and altered for fighting against diseases in a large number of crops using various conventional approaches and biotechnological tools. Identification of R genes and their corresponding Avr genes in many cases played a significant role in understanding of R-Avr gene interactions. Rapid cloning of R genes and editing of susceptible R genes are the other avenues that have broadened the horizon of utilizing R genes in crop improvement programmes. Further, combining R genes with quantitative disease resistance genes has paved the way to develop durable resistance in cultivars. The recent advances in genetics, genomics, bioinformatics and other OMICS tools are now providing greater prospects for deeper understanding of host–pathogen interaction.
This is a preview of subscription content, log in via an institution to check access.
Access this article
Subscribe and save.
- Get 10 units per month
- Download Article/Chapter or eBook
- 1 Unit = 1 Article or 1 Chapter
- Cancel anytime
Price includes VAT (Russian Federation)
Instant access to the full article PDF.
Rent this article via DeepDyve
Institutional subscriptions
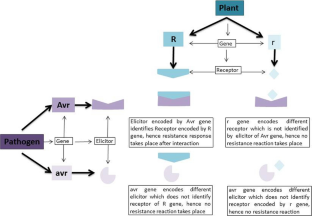
Similar content being viewed by others

Resistance Gene Identification, Cloning, and Characterization in Plants

Genetics and Genomic Approaches for Disease Resistance in Brassicas

Genome engineering of disease susceptibility genes for enhancing resistance in plants
Adugna A. 2004 Alternate approaches for deploying genes for disease resistance in crop plants. Asian. J. Pl. Sci. 3 , 618–623.
Article Google Scholar
Agrios G. N. 1969 Plant pathology , Elsevier Academic Press, UK.
Google Scholar
Ali S., Gladieux P., Leconte M., Gautier A., Justesen A. F., Hovmøller M. S. et al . 2014 Origin, migration routes and worldwide population genetic structure of the wheat yellow rust pathogen Puccinia striiformis f sp tritici. PLoS Pathog. 10 , e1003903.
Article PubMed PubMed Central CAS Google Scholar
Ansan-Melayah D., Balesdent M. H., Delourme R., Pilet M. L., Tanguy X., Renard M. et al . 1998 Genes for race-specific resistance against blackleg disease in Brassica napus L. Plant Breed. 117 , 373–378.
Arora S., Steuernagel B., Gaurav K., Chandramohan S., Long Y., Matny O. et al . 2019 Resistance gene cloning from a wild crop relative by sequence capture and association genetics. Nat. Biotechnol. 372 , 139–143.
Article CAS Google Scholar
Bai Y., Pavan S., Zheng Z., Zappel N. F., Reinstädler A., Lotti C. et al . 2008 Naturally occurring broad-spectrum powdery mildew resistance in a Central American tomato accession is caused by loss of Mlo function. Mol. Plant. Microbe. Interact. 21 , 30–39.
Article CAS PubMed Google Scholar
Bariana H. S. 2003 Breeding for disease resistance. In Encyclopedia of applied plant sciences , pp. 244–253. Harcourt, UK.
Chapter Google Scholar
Bent A. F. and Mackey D. 2007 Elicitors, effectors, and R genes, the new paradigm and a lifetime supply of questions. Annu. Rev. Phytopathol. 45 , 399–436.
Bent A. F., Kunkel B. N., Dahlbeck D., Brown K. L., Schmidt R., Giraudat J. et al . 1994 RPS2 of Arabidopsis thaliana , a leucine-rich repeat class of plant disease resistance genes. Science 265 , 1856–1860.
Bevan J. R., Clarke D. D. and Crute I. R. 1993 Resistance to Erysiphe fischeri in two populations of Senecio vulgaris . Plant. Pathol. 42 , 636–646.
Biffen R. H. 1905 Mendel’s laws of inheritance and wheat breeding. J. Agri. Sci. 1 , 4–8.
Blanvillain-Baufumé S., Reschke M., Sole M., Auguy F., Doucoure H., Szurek B. et al . 2017 Targeted promoter editing for rice resistance to Xanthomonas oryzaea pv. Oryzeae reveals differential activities for SWEET 14 -inducing TAL effectors. Plant Biotechnol. J. 15 , 306–317.
Article PubMed CAS Google Scholar
Browning J. A. and Frey K. J. 1969 Multiline cultivars as a means of disease control. Ann. Rev. Phytopathol. 7 , 355–382.
Ceasar S. A. and Ignacimuthu S. 2012 Genetic engineering of crop plants for fungal resistance: role of antifungal genes. Biotechnol. Lett. 34 , 995–1002.
Center P. G. 1994 The product of the tobacco mosaic virus resistance gene N, similarity to toll and the interleukin-1 receptor. Cell 73 , 1101–1115.
Cesari S., Thilliez G., Ribot C., Chalvon V., Michel C., Jauneau A. et al . 2013 The rice resistance protein pair RGA4/RGA5recognizes the Magnaporthe oryzae effectors AVR-Pia and AVR1-CO39 by direct binding. Plant Cell 25 , 1463–1481.
Article CAS PubMed PubMed Central Google Scholar
Chaijuckam P., Baek J. M., Greer C. A., Webster R. K. and Davis R. M. 2010 Population structure of Rhizoctonia oryzae - sativae in California rice fields. Phytopathol. 100 , 502–510.
Chen R. S., Boeger J. M. and McDonald B. A. 1994 Genetic stability in a population of a plant pathogenic fungus over time. Mol. Ecol. 3 , 209–218.
Chen L. Q., Hou B. H., Lalonde S., Takanaga H., Hartung M. L., Qu X. et al . 2010 Sugar transporters for intercellular exchange and nutrition of pathogens. Nature 468 , 527–532..
Consonni C., Humphry M. E., Hartmann H. A., Livaja M., Durner J., Westphal L. et al . 2006 Conserved requirement for a plant host cell protein in powdery mildew pathogenesis. Nat. Genet. 38 , 716–720.
Croll D. and Laine A. L. 2016 What the population genetic structures of host and pathogen tell us about disease evolution? New Phytologist 212 , 537–539.
Article PubMed Google Scholar
Dadrezaie S. T., Lababidi S., Nazari K., Goltapeh E. M., Afshari F., Alo F. et al . 2013 Molecular genetic diversity in Iranian populations of Puccinia triticina , the causal agent of wheat leaf rust. Am. J. Plant Sci. 4 , 1375.
Dixon M. S., Jones D. A., Keddie J. S., Thomas C. M., Harrison K. and Jones J. D. G. 1996 The tomato Cf-2 disease resistance locus comprises two functional genes encoding leucine-rich repeat proteins. Cell 84 , 451–459.
Dong O. X. and Ronald P. C. 2019 Genetic engineering for disease resistance in plants, recent progress and future perspectives. Plant. Physiol. 180 , 26–38.
Doubly J. A., Flor H. H. and Clagett C. O. 1960 Relation of antigens of Melampsora lini and Linum usitatissimum to resistance and susceptibility. Science 131 , 229–229.
Ellingboe A. H. 1982 Host resistance and host–parasite interactions: A Perspective. In Phytopathogenic prokaryotes , pp. 103-117. Academic Press, New York.
Ellis J. G., Lagudah E. S., Spielmeyer W. and Dodds P. N. 2014 The past, present and future of breeding rust resistant wheat. Front. Plant Sci. 5 , 641.
Article PubMed PubMed Central Google Scholar
Ennos R. A. and McConnell K. C. 1995 Using genetic markers to investigate natural selection in fungal populations. Can. J. Bot. 73 , 302–310.
Felix G., Duran J. D., Volko S. and Boller T. 1999 Plants have a sensitive perception system for the most conserved domain of bacterial flagellin. Plant J. 18 , 265–276.
Flor H. H. 1942a Inheritance of pathogenicity in a cross between physiologic races 22 and 24 of Melampsora lini . Phytopathol. 32 , 5.
Flor H. H. 1942b Inheritance of pathogenicity in Melampsora lini . Phytopathol. 32 , 653–669.
Flor H. H. 1946 Genetics of pathogenicity in Melampsora lini . J. Agric. Res. 73 , 335–357.
Flor H. H. 1955 Host-parasite interaction in flax-rust-its genetics and other implications. Phytopathology 46 , 680–685.
Flor H. H. 1956 The complementary genic systems in flax and flax rust. Adv. Gen. 8 , 29–54.
Flor H. H. and Comstock V. E. 1972 Identification of rust-conditioning genes in flax cultivars. Crop Sci. 12 , 800–804.
Gabriel D. W., Loschke D. C. and Rolfe B. G. 1988 Gene-for-gene recognition, the ion channel defense model. In Molecular genetics of plant-microbe interactions , pp. 3-14. American Phytopathological Society, St. Paul.
Galvez L. C., Banerjee J., Pinar H. and Mitra A. 2014 Engineered plant virus resistance. Plant. Sci. 228 , 11–25.
Gétaz M., Krijger M., Rezzonico F., Smits T. H., van der Wolf J. M. and Pothier J. F. 2018 Genome based population structure analysis of the strawberry plant pathogen Xanthomonas fragariae reveals two distinct groups that evolved independently before its species description. Microb. Genom. 4 , e000189.
PubMed Central Google Scholar
Giannakopoulou A., Steele J. F. C., Segretin M. E., Bozkurt T. O., Zhou J., Robatzek S. et al . 2015 Tomato I2 immune receptor can be engineered to confer partial resistance to the oomycete Phytophthora infestans in addition to the fungus Fusarium oxysporum. Mol. Plant Microbe Interact. 28 , 1316–1329.
Gobbin D., Rumbou A., Linde C. C. and Gessler C. 2006 Population genetic structure of Plasmopara viticola after 125 years of colonization in European vineyards. Mol. Plant Pathol. 7 , 519–531.
Gomez-Casati D. F., Pagani M. A., Busi M. V. and Bhadauria V. 2016 Omics Approaches for the Engineering of Pathogen Resistant Plants. Curr. Issues Mol. Biol. 19 , 89–98.
PubMed Google Scholar
Gómez-Gómez L. and Boller T. 2000 FLS2: an LRR receptor-like kinase involved in the perception of the bacterial elicitor flagellin in Arabidopsis. Mol. Cell 5 , 1003–1011.
Gu K., Yang B., Tian D., Wu L., Wang D., Sreekala C. et al . 2005 R gene expression induced by a type-III effector triggers disease resistance in rice. Nature 435 , 1122–1125.
Halperin S. O., Tou C. J., Wong E. B., Modavi C., Schaffer D. V. and Dueber J. E. 2018 CRISPR-guided DNA polymerases enable diversification of all nucleotides in a tunable window. Nature 560 , 248–252.
Huang N., Angeles E. R. and Domingo J. 1997 Pyramiding of bacterial blight resistance genes in rice, marker assisted selection using RFLP and PCR. Theor. Appl. Genet. 95 , 313–320.
Hückelhoven R. and Panstruga R. 2011 Cell biology of the plantpowdery mildew interaction. Curr. Opin. Plant Biol. 14 , 738–746.
Humphry M., Reinstaedler A., Ivanov S., Bisseling T. O. and Panstruga R. 2011 Durable broad spectrum powdery mildew resistance in pea er1 plants is conferred by natural loss-of function mutations in PsMLO1. Mol. Plant Pathol. 12 , 866–878.
Irzykowska L., Weber Z. and Bocianowski J. 2012 Comparison of Claviceps purpurea populations originated from experimental plots or fields of rye. Open Life Sci. 7 , 839–849.
Ishibashi K., Kezuka Y., Kobayashi C., Kato M., Inoue T., Nonaka T. et al . 2014 Structural basis for the recognition-evasion arms race between Tomato mosaic virus and the resistance gene Tm-1. Proc. Natl. Acad. Sci. USA 111 , E3486–E3495.
Jana T., Sharma T. R. and Singh N. K. 2005 SSR-based detection of genetic variability in the charcoal root rot pathogen Macrophomina phaseolina . Mycol. Res. 109 , 81–86.
Jia H., Orbovic V., Jones J. B. and Wang N. 2016 Modification of the PthA4 effector binding elements in type I CsLOB1 promoter using Cas9/sgRNA to produce transgenic Duncan grapefruit alleviating XccDpthA4: dCsLOB1.3 infection. Plant Biotechnol. J. 14 , 1291–1301.
Jiang W., Zhou H., Bi H., Fromm M., Yang B. and Weeks D. P. 2013 Demonstration of CRISPR/Cas9/sgRNA- mediated targeted gene modification in Arabidopsis, tobacco, sorghum and rice. Nucleic Acids Res. 41 , e188.
Jiménez-Becerril M. F., Hernández-Delgado S., Solís-Oba M. and González Prieto J. M. 2018 Analysis of mitochondrial genetic diversity of Ustilago maydis in Mexico. Mitochondrial DNA Part A 29 , 1–8.
Jiwan D., Roalson E. H., Main D. and Dhingra A. 2013 Antisense expression of peach mildew resistance locus O PpMlo1 gene confers cross-species resistance to powdery mildew in Fragaria x ananassa. Transgenic Res. 22 , 1119–1131.
Johal G. S. and Briggs S. P. 1992 Reductase activity encoded by the HM1 disease resistance gene in maize. Science 258 , 985–987.
Jones J. D. and Dangl J. L. 2006 The plant immune system. Nature 444 , 323–329.
Jones D. A., Thomas C. M., Hammond-Kosack K. E., Balint-Kurti P. J. and Jones J. D. 1994 Isolation of the tomato Cf-9 gene for resistance to Cladosporium fulvum by transposon tagging. Science 266 , 789–793.
JungehÜLsing U. and Tudzynski P. 1997 Analysis of genetic diversity in Claviceps purpurea by RAPD markers. Mycol. Res. 101 , 1–6.
Jupe F., Witek K., Verweij W., Śliwka J., Pritchard L., Etherington G. J. et al . 2013 Resistance gene enrichment sequencing Ren Seq enables reannotation of the NB-LRR gene family from sequenced plant genomes and rapid mapping of resistance loci in segregating populations. Plant J. 76 , 530–544.
Jupe F., Chen X., Verweij W., Witek K., Jones J. D. and Hein I. 2014 Genomic DNA library preparation for resistance gene enrichment and sequencing RenSeq in plants. In Plant-pathogen interactions Humana Press, Totowa, pp. 291–303.
Keen N. T. and Bruegger B. 1977 Phytoalexins and chemicals that elicit their production in plants. In Host plant resistance to pests American Chemical Society, Washington, pp. 1–26.
Keswani C., Bisen K., Singh S. P., Sarma B. K. and Singh H. B. 2016 A proteomic approach to understand the tripartite interactions between plant-Trichoderma-pathogen: investigating the potential for efficient biological control. In Plant, soil and microbes , pp. 79-93. Springer.
Kim S. H., Qi D., Ashfield T., Helm M. and Innes R. W. 2016 Using decoys to expand the recognition specificity of a plant disease resistance protein. Science 351 , 684–687.
Kooman-Gersmann M., Honee G., Bonnema G. and De Wit P. 1996 A high-affinity binding site for the AVR9 peptide elicitor of Cladosporium fulvum is present on plasma membranes of tomato and other solanaceous plants. Plant Cell 8 , 929–938.
Kourelis J. and van der Hoorn R. A. 2018 Defended to the nines, 25 years of resistance gene cloning identifies nine mechanisms for R protein function. Plant Cell 30 , 285–299.
Krasileva K. V., Dahlbeck D. and Staskawicz B. J. 2010 Activation of an Arabidopsis resistance protein is specified by the in planta association of its leucine-rich repeat domain with the cognate oomycete effector. Plant Cell 22 , 2444–2458.
Kroj T., Chanclud E., Michel-Romiti C., Grand X. and Morel J. B. 2016 Integration of decoy domains derived from protein targets of pathogen effectors into plant immune receptors is widespread. New Phytol. 210 , 618–626.
Kumari M., Rai A. K., Devanna B. N., Singh P. K., Kapoor R., Rajashekara H. et al . 2017 Co-transformation mediated stacking of blast resistance genes Pi54 and Pi54rh in rice provides broad spectrum resistance against Magnaporthe oryzae . Plant. Cell. Rep. 36 , 1747–1755.
Lewis J. D., Wu R., Guttman D. S. and Desveaux D. 2010 Allelespecific virulence attenuation of the Pseudomonas syringae HopZ1a type III effector via the Arabidopsis ZAR1 resistance protein. PLoS Genet. 6 , e1000894..
Li T., Liu B., Spalding M. H., Weeks D. P. and Yang B. 2012 High-efficiency TALEN-based gene editing produces disease-resistant rice. Nat. Biotechnol. 30 , 390–392.
Li H., Zhou G. Y., Liu J. A. and Xu J. 2016 Population genetic analyses of the fungal pathogen Colletotrichum fructicola on tea-oil trees in China. PloS One 11 , e0156841.
Liu Q., Yuan M., Zhou Y., Li X., Xiao J. and Wang S. 2011 A paralog of the MtN3/saliva family recessively confers race-specific resistance to Xanthomonas oryzae in rice. Plant Cell Environ. 34 , 1958–1969.
Loegering W. Q. and Ellingboe A. H. 1987 H. H. Flor, Pioneer in phytopathology. Ann. Rev. Phytopathol. 25 , 59–66.
Lorang J. M., Sweat T. A. and Wolpert T. J. 2007 Plant disease susceptibility conferred by a “resistance” gene. Proc. Natl. Acad. Sci. USA 104 , 14861–14866.
Luderer R. 2001 No evidence for binding between resistance gene product Cf-9 of tomato and avirulence gene product AVR9 of Cladosporium fulvum . Mol. Plant Microbe Interact. 14 , 867–876.
Luo Y., Ma T., Zhang A., Ong K. H., Li Z., Yang J. and Yin Z. 2016 Marker-assisted breeding of the rice restorer line Wanhui 6725 for disease resistance, submergence tolerance and aromatic fragrance. Rice 9 , 1–13.
Maciel J. L., Ceresini P. C., Castroagudin V. L., Zala M., Kema G. H. and McDonald B. A. 2014 Population structure and pathotype diversity of the wheat blast pathogen Magnaporthe oryzae 25 years after its emergence in Brazil. Phytopathology 104 , 95–107.
Martin G. B. 1996 Molecular cloning of plant disease resistance genes. In Plant-microbe interactions Springer, Boston, pp. 1–32.
Martin G. B., Brommonschenkel S. H., Chunwongse J., Frary A., Ganal M. W., Spivey R. et al . 1993 Map-based cloning of a protein kinase gene conferring disease resistance in tomato. Science 262 , 1432–1436.
McDonald B. A. 1997 The population genetics of fungi, tools and techniques. Phytopathol. 874 , 448–453.
McDonald B. A. and Linde C. 2002 Pathogen population genetics, evolutionary potential and durable resistance. Ann. Rev. Phytopathol. 40 , 349–379.
McDowell J. M. and Woffenden B. J. 2003 Plant disease resistance genes, recent insights and potential applications. Trends Biotechnol. 21 , 178–183.
Mi J., Yang D., Chen Y., Jiang J., Mou H., Huang J. et al . 2018 Accelerated molecular breeding of a novel P/TGMS line with broad-spectrum resistance to rice blast and bacterial blight in two-line hybrid rice. Rice 11 , 11.
Miedaner T. and Korzun V. 2012 Marker-assisted selection for disease resistance in wheat and barley breeding. Phytopathology 1026 , 560–566.
Mindrinos M., Katagiri F., Yu G. L. and Ausubel F. M. 1994 The A. thaliana disease resistance gene RPS2 encodes a protein containing a nucleotide-binding site and leucine-rich repeats. Cell 78 , 1089–1099.
Moges A. D., Admassu B., Belew D., Yesuf M., Njuguna J., Kyalo M. and Ghimire S. R. 2016 Development of microsatellite markers and analysis of genetic diversity and population structure of Colletotrichum gloeosporioides from Ethiopia. PloS One 11(3) , e0151257.
Mohan K. M., Madhav M. S., Prasad M. S., Devi S. J., Kumar G. R. and Viraktamath B. C. 2012 Analysis of population structure of Magnaporthe grisea using genome specific microsatellite markers. Curr. Trends. Biotechnol. Pharm. 6 , 173–182.
CAS Google Scholar
Mundt C. C. 2002 Use of multiline cultivars and cultivar mixtures for disease management. Ann. Rev. Phytopathol. 40 , 381–410.
Oliva R., Ji C., Atienza-Grande G., Huguet-Tapia J., Prez-Quintero A. and Li T. 2019 Broad-spectrum resistance to bacterial blight in rice using genome editing. Nat. Biotechnol. 37 , 1344–1350.
Onaga G., Wydra K., Koopmann B., Séré Y. and von Tiedemann A. 2015 Population structure, pathogenicity and mating type distribution of Magnaporthe oryzae isolates from East Afr . Phytopathol. 105 , 1137–1145.
Owati A., Agindotan B. and Burrows M. 2019 First microsatellite markers developed and applied for the genetic diversity study and population structure of Didymella pisi associated with ascochyta blight of dry pea in Montana. Fungal Biol. 123 , 384–392.
Panwar V., McCallum B. and Bakkeren G. 2013a Endogenous silencing of Puccinia triticina pathogenicity genes through in planta-expressed sequences leads to the suppression of rust diseases on wheat. Plant J. 73 , 521–948.
Panwar V., McCallum B. and Bakkeren G. 2013b Host-induced gene silencing of wheat leaf rust fungus Puccinia triticina pathogenicity genes mediated by the Barley stripe mosaic virus. Plant Mol. Biol. 81 , 595–608.
Pavan S., Schiavulli A., Appiano M., Marcotrigiano A. R., Cillo F., Visser R. G. F. et al . 2011 Pea powdery mildew er1 resistance is associated to loss-of-function mutations at a MLO homologous locus. Theor. Appl. Genet. 123 , 1425–1431.
Piffanelli P., Zhou F., Casais C., Orme J., Jarosch B., Schaffrath U. et al . 2002 The barley MLO modulator of defense and cell death is responsive to biotic and abiotic stress stimuli. Plant Physiol. 129 , 1076–1085.
Pink D. A. and Hand P. 2003 Plant resistance and strategies for breeding resistant varieties. Plant Protect. Sci. 38 , 9–14.
Pink D. and Puddephat I. 1999 Deployment of disease resistance genes by plant transformation a ‘mix and match’ approach. Trends Plant. Sci. 4 , 71–75.
Pombo M. A., Zheng Y., Fernandez-Pozo N., Dunham D. M., Fei Z. and Martin G. B. 2014 Transcriptomic analysis reveals tomato genes whose expression is induced specifically during effector-triggered immunity and identifies the Epk1 protein kinase which is required for the host response to three bacterial effector proteins. Genome Biol. 15 , 492.
Pradhan S. K., Nayak D. K., Mohanty S., Behera L., Barik S. R., Pandit E. et al . 2015 Pyramiding of three bacterial blight resistance genes for broad-spectrum resistance in deepwater rice variety Jalmagna. Rice 8 , 19.
Article PubMed Central Google Scholar
Prasad P., Savadi S., Bhardwaj S. C. and Gupta P. K. 2020 The progress of leaf rust research in wheat. Fungal Biol. 124 , 537–550.
Pruitt R. N. 2015 The rice immune receptor XA21 recognizes a tyrosine-sulfated protein from a Gram-negative bacterium. Sci. Adv. 1 , e1500245.
Ravensdale M., Bernoux M., Ve T., Kobe B., Thrall P. H., Ellis J. G. and Dodds P. N. 2012 Intramolecular interaction influences binding of the Flax L5 and L6 resistance proteins to their AvrL567 ligands. PLoS Pathog. 8 , e1003004.
Rêgo T. J., Elena G., Correia K. C., Tovar-Pedraza J. M., Câmara M. P., Armengol J. et al . 2019 Genetic diversity and population structure of Lasiodiplodia theobromae from different hosts in northeastern Brazil and Mexico. Plant Pathol. 68 , 930–938.
Römer P., Hahn S., Jordan T., Strauss T., Bonas U. and Lahaye T. 2007 Plant pathogen recognition mediated by promoter activation of the pepper Bs3 resistance gene. Science 318 , 645–648.
Rotblat B., Enshell-Seijffers D., Gershoni J. M., Schuster S. and Avni A. 2002 Identification of an essential component of the elicitation active site of the EIX protein elicitor. Plant J. 32 , 1049–1055.
Sadanand S. 2018 EvolvR-ing to targeted mutagenesis. Nat. Biotechnol. 36 , 819.
Salmeron J. M., Oldroyd G. E. D., Rommens C. M. T., Scofield S. R., Kim H.-S., Lavelle D. T. et al . 1996 Tomato Prf is a member of the leucine-rich repeat class of plant disease resistance genes and lies embedded within the Pto kinase gene cluster. Cell 86 , 123–133.
Sanchez A. C., Brar D. S. and Huang N. 2000 Sequence tagged site marker assisted selection for three bacterial blight resistance genes in rice. Crop Sci. 40 , 792–797.
Sarris P. F., Cevik V., Dagdas G., Jones J. D. G. and Krasileva K. V. 2016 Comparative analysis of plant immune receptor architectures uncovers host proteins likely targeted by pathogens. BMC Biol. 14 , 8.
Seto D., Koulena N., Lo T., Menna A., Guttman D. S. and Desveaux D. 2017 Expanded type III effector recognition by the ZAR1 NLR protein using ZED1-related kinases. Nat. Plant 3 , 17027.
Sharma T. R., Rai A. K., Gupta S. K., Vijayan J., Devanna B. N. and Ray S. 2012 Rice blast management through host-plant resistance: retrospect and prospects. Agric. Res. 1 , 37–52.
Sharma Poudel R., Al-Hashel A. F., Gross T., Gross P. and Brueggeman R. 2018 Pyramiding rpg4 and Rpg1-mediated Stem rust resistance in barley requires the Rrr1 gene for both to function. Front. Plant. Sci. 9 , 1789.
Shen Q.-H., Zhou F., Bieri S., Haizel T., Shirasu K. and SchulzeLefert P. 2003 Recognition specificity and RAR1/SGT1 dependence in barley Mla disease resistance genes to the powdery mildew fungus. Plant Cell 15 , 732–744.
Singh S., Sidhu J. S. and Huang N. 2001 Pyramiding three bacterial blight resistance genes xa5 , xa13 and Xa21 using marker assisted selection into indica cultivar PR106. Theor. Appl. Genet. 102 , 1011–1015.
Steuernagel B., Witek K., Jones J. D. and Wulff B. B. 2017. MutRenSeq, a method for rapid cloning of plant disease resistance genes. In Wheat rust diseases , pp. 215-229. Humana Press, New York.
Strauss T. 2012 RNA-seq pinpoints a Xanthomonas TALeffector activated resistance gene in a large-crop genome. Proc. Natl. Acad. Sci. USA 109 , 19480–19485.
Streubel J., Pesce C., Hutin M., Koebnik R., Boch J. and Szurek B. 2013 Five phylogenetically close rice SWEET genes confer TAL effector-mediated susceptibility to Xanthomonas oryzae pv. oryzae . New. Phytol. 200 , 808–819.
Sundar A. R., Ashwin N. M., Barnabas E. L., Malathi P. and Viswanathan R. 2015 Disease resistance in sugarcane–An overview. Scientia. Agraria. Paranaensis 14 , 200–212.
Tan M. A., Hutten R. C., Visser R. G. and van Eck H. J. 2010 The effect of pyramiding Phytophthora infestans resistance genes R Pi-mcd1 and R Pi-ber in potato. Theo. Appl. Genet. 121 , 117–125.
Tian D., Wang J., Zeng X., Gu K., Qiu C., Yang X. et al . 2014 The rice TAL effector-dependent resistance protein XA10 triggers cell death and calcium depletion in the endoplasmic reticulum. Plant Cell 26 , 497–515.
Truniger V. and Aranda M.A. 2009 Recessive resistance to plant viruses. In Advances in virus research: natural and engineered resistance to plant viruses (ed. G. Loebenstein and J. P. Carr Part), pp. 119–231. Academic Press.
Van Der Biezen E. A. and Jones J. D. 1998 Plant disease-resistance proteins and the gene-for-gene concept. Trends. Biochem. Sci. 23 , 454–456.
van der Hoorn R. A. and Kamoun S. 2008 From guard to decoy: a new model for perception of plant pathogen effectors. Plant Cell 20 , 2009–2017.
Van der Plank J. E. 1968 Disease resistance in plants , pp. 5–7. Academic Press, New York,
Van Schie C. C. and Takken F. L. 2014 Susceptibility genes 101, how to be a good host. Annu. Rev. Phytopathol. 52 , 551–581.
Varallyay E., Giczey G. and Burgyan J. 2012 Virus-induced gene silencing of Mlo genes induces powdery mildew resistance in Triticum aestivum . Arch. Virol. 157 , 1345–1350.
Vlot A. C., Dempsey D. A. and Klessig D. F. 2009 Salicylic acid, a multifaceted hormone to combat disease. Annu. Rev. Phytopathol. 47 , 177–206.
Voytas D. F. and Gao C. 2014 Precision genome engineering and agriculture, opportunities and regulatory challenges. PLoS Biol. 126 , e1001877.
Waltz E. 2018 With a free pass., CRISPR-edited plants reach market in record time. Nat. Biotechnol. 36 , 6–7.
Wang Y., Cheng X., Shan Q., Zhang Y., Liu J. and Gao C. 2014 Simultaneous editing of three homoeoalleles in hexaploid bread wheat confers heritable resistance to powdery mildew. Nat. Biotechnol. 32 , 947.
Wang G., Roux B., Feng F., Guy E., Li L., Li N. et al . 2015a The decoy substrate of a pathogen effector and a pseudokinase specify pathogen-induced modified-self recognition and immunity in plants. Cell Host Microbe 18 , 285–295.
Wang C., Zhang X., Fan Y., Gao Y., Zhu Q., Zheng C. et al . 2015b XA23 is an executor R protein and confers broad-spectrum disease resistance in rice. Mol. Plant 8 , 290–298.
Wheeler H. 1975 Plant pathogenesis , Springer, New York.
Book Google Scholar
Wolfe M. S. and McDermott J. M. 1994 Population genetics of plant pathogen interactions, the example of the Erysiphe graminis-Hordeum vulgare pathosystem. Ann. Rev. Phytopathol. 32 , 89–113.
Wolt J. D., Wang K. and Yang B. 2016 The regulatory status of genome-edited crops. Plant Biotechnol. J. 142 , 510–518.
Wu Y., Xiao N., Chen Y., Yu L., Pan C., Li Y. et al . 2019 Comprehensive evaluation of resistance effects of pyramiding lines with different broad-spectrum resistance genes against Magnaporthe oryzae in rice Oryza sativa L. Rice 12 , 11.
Xiao W., Yang Q., Huang M., Guo T., Liu Y., Wang J. et al . 2019 Improvement of rice blast resistance by developing monogenic lines, two-gene pyramids and three-gene pyramid through MAS. Rice 12 , 78.
Yin L., Zhang Y., Hao Y. and Lu J. 2014 Genetic diversity and population structure of Plasmopara viticola in China. Eur. J. Plant. Pathol. 140 , 365–376.
Zhang K., Halitschke R., Yin C., Liu C. J. and Gan S. S. 2013 Salicylic acid 3-hydroxylase regulates Arabidopsis leaf longevity by mediating salicylic acid catabolism. Proc. Natl. Acad. Sci. USA 110 , 14807–14812.
Zhang X., Peng G., Parks P., Hu B., Li Q., Jiang L. et al . 2017 Identifying seedling and adult plant resistance of Chinese Brassica napus germplasm to Leptosphaeria maculans . Plant Pathol. 665 , 752–762.
Zheng Z., Nonomura T., Appiano M., Pavan S., Matsuda Y., Toyoda H. et al . 2013 Loss of function in Mlo orthologs reduces susceptibility of pepper and tomato to powdery mildew disease caused by Leveillula taurica . PloS One 8 , e70723.
Zhong X., Zhou Q., Cui N., Cai D. and Tang G. 2019 BvcZR3 and BvHs1pro-1 genes pyramiding enhanced beet cyst nematode Heterodera schachtii Schm resistance in oilseed rape Brassica napus L. Int. J. Mol. Sci. 20 , 1740.
Article CAS PubMed Central Google Scholar
Download references
Author information
Authors and affiliations.
Depatment of Plant Breeding and Genetics, Punjab Agricultural University, Ludhiana, 141 004, India
Bhavjot Kaur, Dharminder Bhatia & G. S. Mavi
You can also search for this author in PubMed Google Scholar
Corresponding author
Correspondence to Dharminder Bhatia .
Additional information
Corresponding editor: Manoj Prasad
Rights and permissions
Reprints and permissions
About this article
Kaur, B., Bhatia, D. & Mavi, G.S. Eighty years of gene-for-gene relationship and its applications in identification and utilization of R genes. J Genet 100 , 50 (2021). https://doi.org/10.1007/s12041-021-01300-7
Download citation
Received : 28 June 2020
Revised : 03 March 2021
Accepted : 08 March 2021
Published : 02 July 2021
DOI : https://doi.org/10.1007/s12041-021-01300-7
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
- gene-for-gene hypothesis
- R gene identification and cloning
- R gene classification
- gene pyramiding
- R gene editing.
- Find a journal
- Publish with us
- Track your research

An official website of the United States government
The .gov means it’s official. Federal government websites often end in .gov or .mil. Before sharing sensitive information, make sure you’re on a federal government site.
The site is secure. The https:// ensures that you are connecting to the official website and that any information you provide is encrypted and transmitted securely.
- Publications
- Account settings
Preview improvements coming to the PMC website in October 2024. Learn More or Try it out now .
- Advanced Search
- Journal List
- Philos Trans R Soc Lond B Biol Sci
- v.365(1544); 2010 Apr 27
Mutation and the evolution of recombination
N. h. barton.
1 Institute of Science and Technology, Am Campus 1, A-3400 Klosterneuburg, Austria
2 Institute of Evolutionary Biology, University of Edinburgh, King's Buildings, West Mains Road, Edinburgh EH9 3JT, UK
Under the classical view, selection depends more or less directly on mutation: standing genetic variance is maintained by a balance between selection and mutation, and adaptation is fuelled by new favourable mutations. Recombination is favoured if it breaks negative associations among selected alleles, which interfere with adaptation. Such associations may be generated by negative epistasis, or by random drift (leading to the Hill–Robertson effect). Both deterministic and stochastic explanations depend primarily on the genomic mutation rate, U . This may be large enough to explain high recombination rates in some organisms, but seems unlikely to be so in general. Random drift is a more general source of negative linkage disequilibria, and can cause selection for recombination even in large populations, through the chance loss of new favourable mutations. The rate of species-wide substitutions is much too low to drive this mechanism, but local fluctuations in selection, combined with gene flow, may suffice. These arguments are illustrated by comparing the interaction between good and bad mutations at unlinked loci under the infinitesimal model.
1. Introduction
Mutation is the ultimate source of all genetic variation, and is essential for evolution by natural selection: indeed, most of our genome has been shaped primarily by mutation and random drift. Following the rediscovery of Mendel's laws at the turn of the last century, the first geneticists emphasized major mutations as being responsible for the origin of species. In contrast, the biometricians who were establishing the first statistical studies of evolution emphasized the role of selection in shaping standing variation, by bringing together many slight variations into favourable combinations ( Provine 1971 ). Two decades later, after the efficacy of selection on slight Mendelian variants had been established by both theory and by breeding experiments, different views on the role of mutation in evolution persisted in the contrast between the ‘classical’ and the ‘balance’ views ( Lewontin 1974 ). Under the classical view, associated with Hermann Muller, variation around the wild-type is maintained by a short-term balance between mutation and selection. Thus, standing variation is mere noise, and adaptation is due to favourable mutations—either rare novelties, or alleles that become favourable following a change in environment. On this view, the pattern of standing variation is barely relevant, and adaptation is more or less directly dependent on mutation. In contrast, on the balance view, variation is maintained by complex processes such as overdominance, frequency-dependent selection and heterogeneous selection in structured populations. Although mutation ultimately provides variation, it has little influence on standing variation or on adaptation: a change in mutation rate would, on this view, have little influence.
Understanding the effects of sex and recombination, and why they are so widespread, depends on understanding the nature and causes of mutational variation. It seems most profitable to focus on the role of mutation, taking the classical view, simply because this is theoretically straightforward, and because mutation is a universal process, with a well-known molecular basis, which is open to empirical study using model organisms. In contrast, on the balance view, variation is due to complex interactions between ecological environment and population structure, which are hard to capture in laboratory studies. So, the key question is whether direct effects of good and bad mutations are sufficient to explain the prevalence of sexual reproduction and high rates of recombination. I will focus on eukaryotes with Mendelian inheritance, though many of the same issues arise with bacteria, archaea and viruses.
It seems most likely that sex and recombination evolved, and are maintained, because they generate variation which is the raw material for adaptation by natural selection. This idea was long ago set out by Weismann (1889 ; see Burt 2000 ), but it has taken a considerable theoretical effort to understand it clearly. Other theories exist, in which sex and recombination are side-effects of mechanisms for repairing double-stranded damage to DNA, or gain an advantage by impeding the response to fluctuating epistatic selection, or by reducing competition between siblings ( Williams 1975 ; Bernstein et al. 1988 ; Kondrashov 1993 ; Hamilton 1996 ), However, these seem unlikely to provide a compelling explanation that applies across a broad range of organisms ( Barton & Charlesworth 1998 ; Otto & Lenormand 2002 ; Agrawal 2006 ).
A population genetic advantage to recombination requires high levels of selected polymorphism, so that alleles under selection at different genetic loci have the opportunity to interact, and to be reshuffled by recombination. Recombination cannot be selected unless there are non-random associations between alleles (i.e. linkage disequilibria) that it can break up. Adaptation can be measured by the increase in mean fitness of the population caused by selection on allele frequencies, which is equal to the additive genetic variance in fitness ( Fisher 1930 ). Thus, recombination will increase the rate of adaptation if it increases the additive variance in fitness, and it will only do that if the variance is depressed by negative associations between favourable alleles. In general, then, sex and recombination will be advantageous if there tend to be negative linkage disequilibria between favourable alleles (+ with −, and − with +) that perversely interfere with selection. Moreover, modifiers that increase the rates of sex and recombination will themselves gain a transient advantage through an association with the favourable combinations of alleles that they help to generate. What is crucial, then, is to understand how sufficiently strong and widespread negative linkage disequilibria can arise. Following Felsenstein (1974) , I will distinguish between associations generated by deterministic selection versus random drift; following the theme of this issue, I focus on associations among mutations, good and bad.
Under the classical view, recombination allows deleterious mutations to be eliminated more efficiently, and increases the rate at which favourable alleles can be brought together, despite their association with deleterious alleles. First, I consider the effects of negative linkage disequilibria that are generated deterministically by negative epistasis, and in particular, by truncation selection. I will contrast three cases: asexual reproduction, unlinked loci and most extreme, a population that is forced into linkage equilibrium in every generation. Following Charlesworth ( 1990 , 1993 a ) I will use the infinitesimal model, which neglects changes in allele frequency as being very slow, relative to changes in linkage disequilibrium among loosely linked loci. I then turn to the effects of random linkage disequilibria that are generated stochastically, by sampling drift. Here, considerable progress can be made by following the probability of fixation of a single favourable allele within a very large population, modelled as a branching process. Together, these theoretical models give a rather general understanding of the effects of free recombination, which can be related to observable features of spontaneous mutations.
As will be clear from the reference list, Brian Charlesworth has played a key role in shaping the research described here—both empirical and theoretical. He showed how ‘background selection’ owing to deleterious mutations could explain patterns of neutral diversity in Drosophila ( Charlesworth et al. 1995 ), and the degeneration of non-recombining regions ( Bachtrog & Charlesworth 2002 ; Kaiser & Charlesworth 2008 ; Betancourt et al. 2009 ); how negative epistasis causes selection for sex and recombination (Charlesworth 1990 , 1993 a , b ); and helped give the first direct estimates of genomic mutation rate in Drosophila ( Haag-Liautard et al. 2007 ), and estimates of their effects on fitness ( Loewe & Charlesworth 2006 ).
2. Deterministic associations
In an asexual population, subject to unidirectional deleterious mutation away from the wild-type, at a rate U per genome per generation, the mean fitness is reduced by a factor of exp (− U ) below the maximum possible ( Kimura & Maruyama 1966 ). Remarkably, this classical result is independent of how selection acts. It can be understood by realizing that at equilibrium, each wild-type individual has to produce one wild-type offspring, yet the chance that an offspring escapes any mutation is exp (− U ), assuming a Poisson distribution of numbers of mutations. So, the wild-type must have fitness exp ( U ) higher than the mean fitness, which in the long run is one in an asexual population. This mutation load imposes a constraint on the genome-wide rate, which may have been especially severe in the first reproducing organisms, and is now, for those organisms with the largest functional genomes.
If the effects of different mutations on fitness multiply together, then a sexual population will remain at linkage equilibrium, and so recombination will have no effect: thus, mean fitness will be exp (− U ) regardless of the mode of reproduction or the pattern of genetic linkage. However, negative epistasis together with recombination allows a far higher mutation rate to be tolerated ( Kimura & Maruyama 1966 ). This can be understood using a graphical argument ( figure 1 a ). If the effects of deleterious mutations on fitness increase as their number accumulates (i.e. if there is negative or synergistic epistasis), then the marginal selection on each additional allele can be much higher for a given genetic load, allowing the equilibrium load to be reduced.

(a) Mutation load with truncation selection

It is not obvious how to judge the effect of the mutation load on absolute fitness under truncation selection, because only a fraction θ reproduces, regardless of how much genetic variance is there, or how many mutations have accumulated. Thus, under strict truncation selection there is no clear upper limit to the mutation rate that can be tolerated. I discuss this vexed question below.
(b) The mutational-deterministic hypothesis
Kondrashov (1988) made a forceful argument for the importance of deleterious mutations in driving the evolution of recombination, which did much to promote further research—both theoretical and empirical. Kondrashov ( 1984 , 1988 ) showed by simulation that modifiers of sex and recombination could gain an advantage by alleviating the load, provided that the total mutation rate, U , is large. Crucially, he pointed out that a high genomic mutation rate ( U > l, say) could only be tolerated if there were both negative epistasis and sexual reproduction. Thus, showing that U > 1 would necessarily imply both negative epistasis, and consequently, selection for sex and recombination.
An influential theoretical result states that in a population at equilibrium under selection alone, modifiers that reduce recombination are always favoured; this is known as the reduction principle ( Feldman & Krakauer 1976 ; Feldman et al. 1996 ). For increased recombination to be selected, there must be either change through time (e.g. fluctuating epistasis, or negative associations between alleles that are increasing), or some other force such as mutation or migration, that can counterbalance change in allele frequency owing to directional selection. Indeed, models that include mutation or migration and so allow an equilibrium, have similar consequences for recombination as directional selection alone ( Feldman et al. 1980 ; Lenormand & Otto 2000 ; Martin et al. 2006 ).
Charlesworth (1990) gave an elegant theoretical analysis that showed how Kondrashov's (1988) ‘mutational deterministic’ hypothesis leads to selection for modifiers of sex and recombination. He assumed that the number of deleterious mutations follows an approximately normal distribution, and that log fitness is a quadratic function of this number, so that selection maintains the normal distribution. Assuming that a large number of genes are involved (i.e. the infinitesimal model), allele frequencies change slowly, and recombination modifiers are affected mainly by changes in linkage disequilibrium owing to epistatic selection. Charlesworth (1990) compared three modes of reproduction: asexual; segregation of two non-recombining genomes in a diploid; and sex and recombination with multiple linear chromosomes. His analysis showed that with large U , and with parameters as estimated for larval viability in Drosophila , there could be substantial selection for recombination. However, most of the effect came from segregation, rather than from recombination, making it hard to explain how high recombination rates are maintained.
(c) A general quasi-linkage equilibrium approximation
Barton (1995 a ) gave a general analysis of selection on weak modifiers of recombination, which allowed for arbitrary interactions among multiple sites. The strength of selection for recombination was approximated by assuming that directional selection, s is weak, relative to recombination ( s ≪ r ), and that epistasis between any particular set of genes is very weak ( ϵ ≪ s 2 ). This allows a ‘quasi-linkage equilibrium’ (QLE) approximation, in which the selection for recombination can be related directly to the effects of selection and recombination on the mean and variance of log fitness:

Selection increases mean fitness by precisely the genotypic variance in fitness, which includes both the additive component owing to the marginal effect of each allele, and also the non-additive component, because of epistasis and dominance interactions. Recombination causes an immediate loss of fitness, log( W ), because of the break-up of gene combinations that had been favoured by epistasis, and similarly, segregation causes a loss owing to the break-up of associations between homologous genes in paternal and maternal genomes that had been generated by dominance components of the variance in fitness. If sex and recombination were to destroy all associations, leaving only the effect of changes in allele frequency, then mean fitness would fall back to an increase equal to the additive genetic variance. It is this immediate ‘recombination load’ ( Charlesworth & Barton 1996 ) that drives the ‘reduction principle’. (Note that the recombination load is bounded above by the non-additive genetic variance in fitness).
If there is negative epistasis, then recombination also inflates the additive genetic variance by breaking- up the negative linkage disequilibria amongst favourable alleles, which increases mean fitness in future generations, if selection keeps acting in the same direction. To the extent that the modifier is linked to alleles that will increase under directional selection, it will tend to increase with them; this is expressed by the first term in equation ( 2.1 ), which involves the reciprocal of the harmonic mean recombination between the modifier and the selected loci. This QLE approximation describes Charlesworth's (1990) analysis well, though the latter extends to cover stronger selection. (Charlesworth's analysis is based primarily on the normal approximation, and does not require weak selection as such). Equation ( 2.1 ) also approximates Charlesworth's (1993 a , b ) analyses of directional and fluctuating selection, which give a similar advantage to recombination as does a mutation-selection balance. To summarize: equation ( 2.1 ) shows that the advantage of recombination depends primarily on how much negative linkage disequilibria reduce the variance in log fitness ( V ).
(d) Difficulties with negative epsitasis

A related difficulty is that it is hard to see why epistasis should tend to be systematically negative. It is true that negative epistasis (in the limit, truncation selection) tends to alleviate various kinds of genetic load ( Kimura & Maruyama 1966 ; Sved 1968 ), but it is not at all clear that it should evolve to be negative. Metabolic models give mixed results, with no clear indication that they would cause negative epistasis ( Keightley & Kacser 1987 ; Szathmary 1993 ). Selection for robustness to environmental and genetic perturbations may lead to negative epistasis, by analogy with arguments for the evolution of dominance: if some ‘safety margin’ has evolved, then moderate loss of function may have little effect on fitness, whereas larger numbers of deleterious mutations, especially when homozygous, may cause a substantial loss of fitness. However, though negative epistasis may evolve in this way in some models, there is again no clear theoretical support for its generality ( Hansen 2006 ).
(e) The cost of selection
As well as reducing the mutation load, recombination with negative epistasis also reduces the ‘cost of natural selection’. Haldane (1957) showed that the total loss of reproductive capacity required to raise an allele from a low frequency p 0 is approximately log (1/ p 0 ); just as with mutation load, this result applies to asexuals and to sexual populations at linkage equilibrium, for any pattern of selection. (Of course, favourable mutations are not in themselves costly: the ‘cost’ is in slow evolution by natural selection, rather than instantaneous adaptation). However, negative epistasis allows any number of rare alleles to be fixed in a sexual population. This can be understood by thinking of the most favourable case of truncation selection. Then, any number of rare alleles can be picked up, and will increase in frequency by a factor 1/ θ in each generation, where θ is the fraction selected. Once these alleles become common, recombination is needed to bring them together and fix the fittest genotype. However, since most of the cost of selection is incurred during the long time that favourable alleles are increasing from low frequency (approx. (1/ S )log(1/ p 0 ) generations), this argument shows that almost all of the cost of selection can be avoided in a freely recombining population. Charlesworth (1993 a ) showed this deterministic advantage of recombination in the less extreme case of directional selection on a quantitative trait that also experiences stabilizing selection towards a moving optimum, and hence, negative epistasis (see also Burger (1999) and Waxman & Peck (1999) ). Here, stabilizing selection reduces the additive genetic variance, and recombination restores it, hence speeding the response.
Recombination also speeds up the response to directional selection in the presence of a mutation load, provided that there are negative interactions between the genes involved. In an infinite population, the response to directional selection on an additive trait is independent of mutation load, if effects on log fitness add up. However, if truncation selection acts on the trait, plus some measure of the mutation load, then negative associations will build up that interfere with selection—individuals with higher trait values will tend to carry a higher load of deleterious mutations, because these will have been partially shielded from selection by the higher trait ( figure 2 ).

The association between a selected trait and the mutation load slows down the response to truncation selection. The upper line shows the response to truncation selection under the infinitesimal model of a trait with genetic variance at linkage equilibrium V 1 = 20; 20 per cent survive in each generation, and linkage disequilibria reduce the variance to 11.6. The lower line shows the response when truncation selection acts on the sum of this trait, and the number of deleterious mutations. Because truncation selection is spread over two traits, and because there is a correlation of 21 per cent between mutation load and the favoured trait, the selection response is substantially reduced ( U = 10, as in figure 1 ).
3. Random associations
(a) reduced diversity in regions of reduced recombination.
About 20 years ago, it was observed that regions of the Drosophila genome with low recombination have low nucleotide diversity at silent sites. This is not associated with any reduction in between-species divergence, and so cannot be explained by differences in mutation rate ( Aguade et al. 1989 ; Stephan & Langley 1989 ; Begun & Aquadro 1992 ). Similar associations have been found in other groups ( Nachman 2002 ), though the causes of the correlation seen in humans remain unclear ( Hellman et al. 2005 ; Spencer et al. 2006 ; Cai et al. 2009 ). Similarly, exceptionally low diversity is seen in genomes, or regions of genome, with little or no recombination, such as Y chromosomes ( Bachtrog & Charlesworth 2002 ; Charlesworth et al. 2009 ), dot chromosomes ( Betancourt et al. 2009 ), obligate selfers ( Charlesworth 2003 ) or endosymbiotic bacteria ( Funk et al. 2001 ).
If such patterns are because of the population genetic effects of recombination, then they must be caused by selection at linked loci, and mediated by linkage disequilibria between selected loci, and the observed neutral markers. Moreover, such linkage disequilibria must be generated by random drift, since there can be no epistasis with neutral markers. Random associations that reduce diversity must also interfere with selection, reducing both the ability of populations to accumulate favourable mutations and to eliminate deleterious ones. This must lead, finally, to selection for modifiers that increase recombination. So, the simple observation of a correlation between neutral diversity and recombination implies the existence of interference among selected loci that must lead to selection for recombination. Here, I summarize the relevant theory, and in §4, return to the interpretation of the relation between diversity and recombination.
(b) The Hill–Robertson effect
As well as producing random fluctuations in allele frequency that reduce genetic diversity, genetic drift also produces random associations between alleles at different loci. These random linkage disequilibria tend to become negative, and so interfere with selection; this interference favours increased recombination, in exactly the same way as when negative linkage disequilibria are generated by epistasis (equation 2.1 ). It seems at first paradoxical that random drift should lead to negative associations between favoured alleles, because the immediate effect of drift is impartial: associations between any pair of alleles are, on average, zero and have the same distribution regardless of how the alleles affect fitness. Thus, the tendency for random associations between alleles to become negative is because of an interaction between drift and selection at the two loci. This can be understood in two ways. First, positive associations will accelerate selection, and will rapidly fix the fit ++ combinations that they produce. In contrast, negative associations shield alleles from selection, reducing the variance in fitness, and so will tend to persist. In the extreme case of asexuality, populations can fix for a mixture of +− and −+ combinations with the same fitness, and so will maintain negative linkage disequilibria indefinitely.
Another way to understand why random linkage disequilibria tend to interfere with directional selection was set out by Hill & Robertson (1966) . The effect on fitness of alleles at one locus are obscured by association with random genetic backgrounds, each with their own effect on fitness. In other words, random association of one locus with other selected loci induce random perturbations that act in the same way as classical random drift, and interfere with selection. On this view, the fluctuations in allele frequency at the selected locus do average to zero, but their long-term effect is negative. To see this, think of a favourable mutation that increases relative fitness by s , and that starts at some low frequency p 0 = 1/2 N . Its chance of fixation is 2s( N e / N ), and so its expected frequency in the long term is 2s( N e / N ) = (4 N e s ) p 0 . Any reduction in the effective population size, N e , reduces its long-term expected frequency in direct proportion; this effect is mediated by negative linkage disequilibria that arise during its passage to fixation, but the effect is most easily understood as an inflation of drift at the focal locus.
(c) Unlinked loci

(d) Asexual populations

Selective sweeps through an asexual population also have a drastic effect, causing ‘periodic selection’ in which all variation is eliminated when a single favourable mutation fixes. Neutral diversity can only build up over the time since the most recent sweep (approx. 1/ Λ ), and so average pairwise diversity is π ∼ 2 μ / Λ . As Fisher (1930) and Muller (1932) first pointed out, advantageous alleles can only fix if they arise within a background that is already on its way to fixation—unless they themselves have an advantage that is large enough to out-compete a previously established selective sweep. Either way, complete linkage drastically reduces the efficiency with which selection can accumulate adaptive mutations (see Rouzine et al. 2008 , for a summary of recent theory for asexual populations).
(e) A linear map

Maynard Smith & Haigh (1974) showed that a selective sweep of strength S reduces neutral diversity at a linked locus by (on average) 2 N ( S ) −2 r / S (see Stephan et al. 1992 ). This can be interpreted as the chance that two lineages coalesce in the unique genome that carried the original positive mutation, rather than recombining away to some more distant ancestry ( figure 3 ). The net rate of coalescence between two lineages because of the rate of recurrent sweeps, Λ , scattered over a map of length R , averages 2( Λ / R ) ( S /log(2 N S )), compared with a rate 1/(2 N ) due to a sampling drift. Thus, neutral diversity is reduced by a factor:

which depends on both the density of sweeps ( Λ / R ) and on the strength of selection relative to drift (2 N S ).

The different effects of a selective sweep on neutral diversity ( a ) and on a weakly favoured allele ( b ). Neutral lineages will only coalesce if they trace right back to near the origin of the sweep. Diagram ( a ) shows two lineages (black, grey) that both trace back into the fitter background, but both then recombine away into the ancestral background, and so remain unrelated. Such recombination, allowing the lineages to escape coalescence, can occur throughout the long time taken for the new mutation to increase from one copy (shown by disc at lower left). Diagram ( b ) shows how a weakly favoured allele is knocked back by a sweep. To survive, it must recombine onto the new background doing the brief duration of the sweep—giving less scope for recombination than for neutral diversity.
A linked selective sweep has a more severe effect on the survival of an advantageous mutation than it does on neutral diversity. While a rare allele with some small advantage, s , is struggling to increase from low frequency, it is vulnerable to being knocked back by the substitution of a strongly selected allele at a linked locus: the effect is as if its frequency were suddenly reduced by a random factor, which averages 1 − ( s / S ) r / S ( Barton 1995 b ), and which will be substantial if linkage is tighter than the advantage of the strongly selected allele ( r ≪ S ). (Throughout this section, s refers to the advantage of the allele that is increasing from low numbers, while S refers to the selection on sweeps that are already established.) In contrast, the effect on neutral diversity is restricted to a narrower region of the genetic map: as we trace a lineage back through the sweep, it can recombine away, onto the ancestral background, at any time back until the sweeping allele originated ( t ∼ (1/ S ) log(4 N e S )) ( figure 3 ). Therefore, the effect of sweeps on neutral diversity is significant over a map length of r ∼ log(4 N e S ), which is much less than r ∼ S for strongly selected sweeps (log(4 N e S ) ≫ 1).
(f) Multiple sweeps
The expected effect of multiple selective sweeps, occurring at random times and at random locations on the genetic map, can be found by approximating their effect as a series of random catastrophes that each reduce allele frequency by some fraction, averaging ( s / S ) − r / S . On average, these will knock back any rare allele at a rate s crit , and so its chance of fixation is just 2( s − s crit ). This result seems puzzling at first, because random hitch-hiking events do not alter the expected frequency of an allele. However, almost all sweeps originate on the common background, and so knock the rare allele back. These are countered by extremely rare events where the favourable mutation arises in coupling with the rare allele, and give it an extremely large boost. However, such rare events can do no more than making fixation certain, and so overall, have negligible effect on fixation probability. Thus, selective sweeps set a threshold selection coefficient, below which an adaptive allele has negligible chance of fixation in a large population. (Strictly speaking, the probability of fixation tends to zero as N tends to infinity, if s < s crit ; Barton 1994 ). This critical threshold is proportional to the variance in log fitness due owing to sweeps, per unit map length ( v / R ; Barton 1994 ):

(g) Interference due to weakly selected alleles
These results assume that the alleles that cause Hill–Robertson interference (whether selected positively or negatively) evolve deterministically. Then, neutral diversity can be found using the structured coalescent ( Wakeley 2008 ), in which lineages trace back through different genetic backgrounds, whose frequencies change in a known way. Similarly, multitype branching processes give the probability of fixation of a single favourable allele, which depends on the genetic background in which it finds itself. What if drift and selection have comparable strength ( N e s ∼ 1), so that the genetic backgrounds responsible for interference fluctuate randomly?
The effect of weakly selected alleles on neutral diversity can be found using the structured coalescent, but allowing for the fluctuating frequencies of the genetic backgrounds ( Hudson & Kaplan 1988 ; Barton & Etheridge 2004 ). If these fluctuations mirror those due purely to drift (as when the backgrounds are defined by neutral alleles), there can be no effect on linked loci. If they fluctuate less (as with balanced polymorphism), then neutral diversity increases, though only in a narrow region of the map ( r ∼ μ ; Hudson & Kaplan 1988 ; Kaplan et al. 1988 ). Conversely, if background frequencies change systematically, as with selective sweeps or deleterious mutations, diversity is reduced. When selection is weak ( N e s ∼ 1), random fluctuations due to drift greatly reduce hitch-hiking effects; for example, the effect of background selection with no recombination is roughly halved when N e s ∼ 3, compared with the large N e s limit ( Barton & Etheridge 2004 ; fig. 12). Nevertheless, because a very large number of sites may be under weak selection, their cumulative effects can be significant ( McVean & Charlesworth 2000 ).
The main outstanding theoretical problem is to understand how selection over a large number of loci generates Hill–Robertson interference, and how that in turn selects for recombination. The infinitesimal model provides a simple approximation for unlinked loci that identifies the additive variance in fitness as the key parameter. However, linked loci can have a much stronger effect—especially, on selection for recombination, since modifiers must remain linked to the fitter combinations that they help produce. The selection for recombination owing to two selected loci can be found theoretically, for both fluctuations in established polymorphisms ( Barton & Otto 2005 ), and for the stochastic increase of a single favourable mutation ( Roze & Barton 2006 ). In both cases, simple extrapolation from two selected loci to many implies that selection for recombination should be very weak—proportional to the square of the heritable fitness variance—unless that variance is very high. Yet, simulations of large numbers of loci show much stronger effects than expected by extrapolation from two selected loci. Iles et al. (2003) simulate selection on standing variation, and show that for fixed variance in fitness, Hill–Robertson interference, and the consequent selection for recombination, increases with the number of loci. Keightley & Otto (2006) simulate deleterious mutation at many loci, and show a similar increase in Hill–Robertson interference with the number of genes. The challenge is to find a theoretical approximation that can explain these patterns.
4. Discussion
This condensed summary of the theory relating to interference between selected loci, and its consequences for the evolution of recombination shows the considerable progress that has been made in understanding the theoretical issues—primarily, in laying out a taxonomy of the distinct issues, and in identifying the importance of key parameters such as the density of mutations and of selective sweeps on the genetic map. In the following discussion, I focus on two issues: the limits to the amount of selection that may be acting, and the evidence as to its actual extent.
(a) Evidence on the extent and consequences of Hill–Robertson interference
The correlation between recombination and diversity, first seen in Drosophila , has driven a large research program—both empirical and theoretical—that aims to answer (at least) four questions. What kind of selection is responsible for reducing neutral diversity? Does this also interfere with selection itself, reducing adaptation as well as neutral diversity in region of low recombination? Are such effects also important across the bulk of the genome, in regions of typical recombination? What is the net selection for recombination?
Most attention has been given to finding whether reduced diversity is due mainly to the flux of favourable mutations, sweeping through to fixation, or to background selection, owing to elimination of deleterious mutations. These have the advantage of providing clear alternative hypotheses, described by simple and observable parameters: the rate of species-wide selective sweeps, Λ , and the genomic mutation rate, U . However, as discussed below, neither are likely to be sufficient explanations, either for reduced diversity and adaptation in regions of low recombination, or for the maintenance of recombination. Selection that fluctuates in time and space may be more important, but cannot be summarized by a few simple parameters. A distinct aspect of this question is whether weakly selected loci ( N e s ∼ 1) have a significant influence on linked loci; such alleles have distinct effects that are harder to analyse or to measure than those evolving deterministically with negligible influence from random drift.
To a first approximation, a reduction in neutral diversity can be seen as due to an increased rate of random drift, described by a reduced effective population size, N e . This is expected to reduce the chance of fixation of favourable alleles, by a ratio N e / N , and to increase the chance that deleterious alleles will fix if N e s is small. However, as explained above, random linkage disequilibria can have substantially different effects on selected alleles than on neutral: on the one hand, fixation probability of favourable mutations can be reduced much more than neutral diversity, but on the other hand, linkage to weakly selected alleles can have much smaller effect than linkage to strongly selected alleles.
Evidence that linkage to selected loci reduces adaptation as well as diversity comes from weaker codon usage bias in regions of low recombination ( Kliman & Hey 1993 ), and from lower rates of non-synonymous substitution and higher frequencies of rare (presumably deleterious) alleles in regions of low recombination ( Betancourt & Presgraves 2002 ; Presgraves 2005 ; Betancourt et al. 2009 ). If Hill–Robertson interference is extensive, then there should be a positive correlation between levels of neutral diversity and the rate of adaptive substitution. However, Macpherson et al. (2007) found that in regions of the Drosophila simulans genome with a higher rate of non-synonymous divergence from D. melanogaster , silent-site diversity is both lower and more heterogeneous and that diversity is also more heterogeneous—consistent with the effect of selective sweeps. Similarly, Cai et al. (2009) found that silent-site diversity is lower in regions with higher divergence and functional density, and with lower recombination. On the other hand, Bullaughey et al. (2008) found no correlation between recombination and non-synonymous divergence in humans. Generally, interpreting correlations between rates of amino acid divergence and recombination is difficult: for example, hominids show a higher rate of divergence in gene expression and in 5′-noncoding sequences than murids, which has been interpreted as owing to accumulation of weakly deleterious substitutions as a result of a lower hominid effective population size ( Keightley et al. 2005 ). By a similar argument, a higher rate of non-synonymous divergence could be seen as being due to Hill–Robertson interference, rather than as causing it, as assumed above: the direction of causation depends on the distribution of selection coefficients.
The correlation between neutral diversity and recombination that is seen in Drosophila does not directly show whether interference from linked loci is significant across the bulk of the genome, in regions of high recombination as well as low. However, it does demonstrate the existence of a source of random drift that could be the main process that shapes neutral variation, and that limits the effectiveness of selection across the whole genome. The key observation that even the most abundant species have only moderately high genetic variation ( Lewontin 1974 ; Nevo et al. 1984 ; Lynch & Conery 2000 ) shows that random drift cannot be simply due to sampling, which would give a negligible rate inversely proportional to census numbers, ∼1/ N . In their original analysis of hitch-hiking, Maynard Smith & Haigh (1974) argued that selective sweeps must necessarily be the dominant source of drift in any sufficiently large population; Gillespie ( 2000 , 2001 ) has elaborated this view, that random drift is primarily due to fixation of favourable mutations. The observation of reduced silent-site diversity in regions of low recombination is consistent with this, and if it is explained by selective sweeps, then Maynard Smith & Haigh's (1974) argument implies that it must be the main source of drift in abundant species. However, there are two caveats. First, background selection reduces effective population size by a constant factor, independent of actual numbers, and so would have the same proportionate effect, however large the population. Second, diversity in abundant species may be limited by sporadic bottlenecks, rather than by selective sweeps. Thus, the two observations of a correlation between diversity and recombination, and of modest diversity in even abundant species could be explained either by a predominant effect of selective sweeps, or by a combination of background selection with population bottlenecks ( figure 4 )—or, of course, by some combination of these three processes.

The relation between neutral diversity, recombination rate R , and population size (small, medium, large, reading upwards), for population ( a ) bottlenecks, ( b ) deleterious mutation and ( c ) selective sweeps. With bottlenecks ( a ), diversity is independent of recombination rate, but reaches an upper limit as census numbers increase. With ‘background selection’ ( b ), diversity increases with recombination but is strictly proportional to census numbers. With selective sweeps ( c ), diversity increases to an upper limit with both population size and recombination.
If selection on linked loci does reduce neutral diversity across the whole genome, then it must also interfere to some extent with selection, and hence must lead to some selection for sex and recombination. The problem is to find whether such selection is strong enough to outweigh the various costs. We now have direct estimates of the total rate of mutation ( Haag-Liautard et al. 2007 ; Lynch et al. 2008 ; Keightley et al. 2009 ); of the distribution of their negative effects on fitness ( Loewe & Charlesworth 2006 ); of the total rate of adaptive species-wide substitutions ( Smith & Eyre-Walker 2002 ; Eyre-Walker & Keightley 2009 ); and (very roughly) of the strength of positive selection involved, inferred from the size of genomic regions of reduced diversity ( Macpherson et al. 2007 ). These estimates are uncertain, largely because of the confounding effect of different kinds of selection, and of population structure. But, leaving these uncertainties aside, would knowledge of such global parameters be enough to tell us the strength of selection for recombination, through some simple relation such as equation ( 2.1 ), which applies to the effects of the linkage disequilibria built up deterministically by epistasis?
A consistent excess of divergence, relative to that expected from within-species polymorphism, indicates that around 30–50 per cent of amino acid substitutions in Drosophila were adaptive; estimates for humans are lower, but might also be substantial ( Eyre-Walker & Keightley 2009 ). Moreover, a substantial fraction of divergence in non-coding regions may also have been adaptive ( Halligan & Keightley 2006 ). The detection of selective sweeps via regions of reduced diversity is consistent with these estimates, and indicates that many substitutions are quite strongly selected—with s ∼ 1 per cent, say ( Macpherson et al. 2007 ); the very fact that so many sweeps can be detected shows that they cause a substantial reduction in diversity (though, with the caveat that a complex demography may give false indications of selection). However, even taking the higher estimates, the overall rate of substitution is so slow that the density of sweeps per map length, Λ / R cannot be very high—certainly, far too low to cause significant selection for recombination via Hill–Robertson interference ( Roze & Barton 2006 ). Estimates of genome-wide mutation rate are more encouraging: a direct estimate of the total rate of deleterious mutation over the diploid genome of D. melanogaster of U ∼ 1.2 ( Haag-Liautard et al. 2007 ) would be enough to give substantial selection for recombination if epistasis is generally negative ( Charlesworth 1990 ). However, while another direct estimate for yeast is also surprisingly high ( Lynch et al. 2008 ), there it seems that the mutations involved are very weakly selected, and so may have little effect on recombination.
Neither deleterious mutations nor the fixation of favourable mutations through the whole species can contribute much heritable variance in fitness—specifically, not enough to cause much interference from unlinked loci. However, it is plausible that the heritable variance is high, as a result of fluctuating selection and local sweeps. If it is high enough for unlinked loci to cause significant interference, then linked loci may give a still larger contribution, though the theory here is undeveloped. The contrast between the ‘classical’ and the ‘balance’ views remains unresolved: it remains to be seen whether the mass of genomic data will tell us about locally fluctuating selection, or will remain limited to estimating global parameters.
(b) Limits to the genetic load
The neutral theory of molecular evolution was motivated by arguments that selection could not act on the whole genome: organisms could not have enough excess reproduction to eliminate deleterious mutations, to maintain balanced polymorphisms, and to fix adaptive substitutions, at an extremely large number of sites ( Kimura 1968 ; King & Jukes 1969 ). Such arguments, framed in terms of various kinds of ‘genetic loads’, have been neglected since the 1970s, when it was shown that truncation selection on a sexual population allows selection to act much more efficiently ( Sved et al. 1967 ; Sved 1968 ). Yet, we must still ask whether real organisms are likely to be selected in this way. What are the highest rates of deleterious mutation, and of adaptive substitution, that can be sustained by a freely recombining population?

How might selection act on real organisms? There are two distinct issues: the relation between individual genotype and relative fitness (i.e. epistasis), and the effect of genotype on absolute fitness. Negative epistasis (in the extreme, truncation selection) reduces the mutation load by allowing much stronger marginal selection on each allele compared with what would be expected from the fitness of the optimal genotype ( figure 1 ). In principle, we could measure the marginal effect of each of the deleterious alleles carried by an individual, and from this, predict the fitness of the ideal genotype by multiplying the marginal effects together. Theory predicts that at linkage equilibrium, this fitness must equal e U , relative to the mean fitness of one for a stable population. Plainly, the actual fitness of the optimal genotype is limited, implying that there must be negative epistasis if U is large ( figure 1 a ; Kondrashov 1988 ). Is it plausible that marginal selection coefficients are in fact as large as is implied by this limit?
Negative epistasis could be owing to extensive redundancy, such that there must be many deleterious mutations before the organism degrades appreciably. This is suggested by carcinogenesis, which typically require multiple defects in the control of the cell cycle, and is the striking fact that a majority of genes in most eukaryotes can be deleted with little phenotypic effect. Yet, on this view, individuals must typically have already accumulated many defects, so that the marginal effect of an extra one is severe; then, a further increase in mutation rate would cause a disproportionate loss of fitness ( figure 1 b ). Thus, to be plausible there must be some mechanism that would shift the fitness curve as the mutation rate changed, or that would limit the mutation rate itself. Under truncation selection, of course, there has to be some feedback such that a fixed fraction survives.
This brings us to the second issue of the relation between genotype and absolute fitness. Roughly, we can think of components of fitness that are required for individual survival and reproduction, regardless of the state of the rest of the population—development to adulthood, survival, fertility and so on. The total rate of mutation to genes involved in these components may be large only if the population evolves to have negative epistasis, such that each mutation has a large marginal effect. There may be other components of fitness that depend on competition between individuals, and do not alter the average number of offspring: for example, male secondary sexual traits, or female preferences for them. These could be under at least approximate truncation selection, and could sustain a high mutation rate. However, it seems likely that the sets of genes that affect the two kinds of trait would largely overlap, so that we cannot just add up the mutation rates for the two. If the effects of shared alleles on the two components of fitness are positively correlated, this will increase the strength of selection on them, and will reduce the load on the first component, that is under ‘hard’ selection ( Agrawal 2001 ). (On the other hand, with negative correlations, so that sexual and natural selection are opposed, the first component of fitness may be depressed even with no mutation.) To raise yet further complications, female preferences may evolve for ‘good genes’ that are associated with increased male vigour. One way by which such preferences might evolve is through an epistatic handicap, in which only the most vigorous males can bear the cost of a signal trait. However, this leads to a positive correlation between fitness components that may increase mutation load.
We are left, then, with a theoretical upper limit under which truncation selection could allow extremely high mutation rates and (by similar arguments) rates of substitution. However, there seems to be no compelling reason why selection should evolve to act in such an efficient way, and so the traditional load arguments retain some force. Recent direct estimates of mutation rate and of the fraction of genome that is constrained by selection suggest that some species (including our own) may suffer a substantial mutation load, sufficient to cause significant selection for recombination ( Charlesworth 1990 ). However, the rate of species-wide substitution in natural populations is too low to cause strong selection for recombination. Nevertheless, it remains possible that local populations experience far more directional selection, and that it is this which sustains widespread sex and recombination.
Acknowledgments
I would like to thank W. G. Hill and L. Loewe for organizing this special issue, and the Royal Society and Wolfson Foundation for their support. Also, A. Kondrashov and L. Loewe gave very helpful comments that helped improve the manuscript.
One contribution of 16 to a Theme Issue ‘The population genetics of mutations: good, bad and indifferent’ dedicated to Brian Charlesworth on his 65th birthday .
- Agrawal A. F.2001 Sexual selection and the maintenance of sexual reproduction . Nature 411 , 692–695 ( doi:10.1038/35079590 ) [ PubMed ] [ Google Scholar ]
- Agrawal A. F.2006 Evolution of sex: why do organisms shuffle their genotypes? Curr. Biol. 16 , 696–704 ( doi:10.1016/j.cub.2006.07.063 ) [ PubMed ] [ Google Scholar ]
- Aguade M., Miyashita N., Langley C. H.1989 Reduced variation in the yellow-achaete-scute region in natural populations of Drosophila melanogaster . Genetics 122 , 607–615 [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Bachtrog D., Charlesworth B.2002 Reduced adaptation of a non-recombining neo-Y chromosome . Nature 416 , 323–326 ( doi:10.1038/416323a ) [ PubMed ] [ Google Scholar ]
- Barton N. H.1994 The reduction in fixation probability caused by substitutions at linked loci . Genet. Res. 64 , 199–208 ( doi:10.1017/S0016672300032857 ) [ Google Scholar ]
- Barton N. H.1995a A general model for the evolution of recombination . Genet. Res. 65 , 123–144 ( doi:10.1017/S0016672300033140 ) [ PubMed ] [ Google Scholar ]
- Barton N. H.1995b Linkage and the limits to natural selection . Genetics 140 , 821–841 [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Barton N. H. Why sex and recombination? Cold Spring Harbor Symp. Quant. Biol. 2009; 74 ( doi:10.1101/sqb.2009.74.030 ) [ PubMed ] [ Google Scholar ]
- Barton N. H., Charlesworth B.1998 Why sex and recombination? Science 281 , 1986–1990 ( doi:10.1126/science.281.5385.1986 ) [ PubMed ] [ Google Scholar ]
- Barton N. H., Etheridge A. M.2004 The effect of selection on genealogies . Genetics 166 , 1115–1131 ( doi:10.1534/genetics.166.2.1115 ) [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Barton N. H., Otto S. P.2005 Evolution of recombination due to random drift . Genetics 169 , 2353–2370 ( doi:10.1534/genetics.104.032821 ) [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Begun D. J., Aquadro C. F.1992 Levels of naturally occurring DNA polymorphism correlate with recombination rate in Drosophila melanogaster . Nature 356 , 519–520 ( doi:10.1038/356519a0 ) [ PubMed ] [ Google Scholar ]
- Bernstein H., Hopf F. A., Michod R. E.1988 Is meiotic recombination an adaptation for repairing DNA, producing genetic variation, or both? In The evolution of sex (eds Michod R. E., Levin B. R.), pp. 139–160 Sunderland, MA: Sinauer Press [ Google Scholar ]
- Betancourt A. J., Presgraves D. C.2002 Linkage limits the power of natural selection in Drosophila . Proc. Natl Acad. Sci. USA 99 , 13 616–13 620 ( doi:10.1073/pnas.212277199 ) [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Betancourt A. J., Welch J. J., Charlesworth B.2009 Reduced effectiveness of selection caused by a lack of recombination . Curr. Biol. 19 , 655–660 ( doi:10.1016/j.cub.2009.02.039 ) [ PubMed ] [ Google Scholar ]
- Bullaughey K., Przeworski M., Coop G.2008 No effect of recombination on the efficacy of natural selection in primates . Genome Res. 18 , 544–554 ( doi:10.1101/gr.071548.107 ) [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Bulmer M. G.1980 The mathematical theory of quantitative genetics New York, NY: Oxford University Press [ Google Scholar ]
- Burger R.1999 Evolution of genetic variability and the advantage of sex and recombination in changing environments . Genetics 153 , 1055–1069 [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Burt A.2000 Sex, recombination and the efficacy of selection—was Weissman right? Evolution 54 , 337–351 [ PubMed ] [ Google Scholar ]
- Cai J. J., Macpherson J. M., Sella G., Petrov D. A.2009 Pervasive hitchhiking at coding and regulatory sites in humans . PLoS Genet. 5 , e1000336. [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Charlesworth B.1990 Mutation-selection balance and the evolutionary advantage of sex and recombination . Genet. Res. 55 , 199–221 ( doi:10.1017/S0016672300025532 ) [ PubMed ] [ Google Scholar ]
- Charlesworth B.1993a Directional selection and the evolution of sex and recombination . Genet. Res. 61 , 205–224 ( doi:10.1017/S0016672300031372 ) [ PubMed ] [ Google Scholar ]
- Charlesworth B.1993b The evolution of sex and recombination in a varying environment . J. Hered. 84 , 345–350 [ PubMed ] [ Google Scholar ]
- Charlesworth B., Barton N. H.1996 Recombination load associated with selection for increased recombination . Genet. Res. 67 , 27–41 ( doi:10.1017/S0016672300033450 ) [ PubMed ] [ Google Scholar ]
- Charlesworth B., Betancourt A. J., Kaiser V. B., Gordo I.2009 Genetic recombination and molecular evolution . Cold Spring Harbor Symp. Quant. Biol. 74 ( doi:10.1101/sqb.2009.74.015 ) [ PubMed ] [ Google Scholar ]
- Charlesworth D.2003 Effects of inbreeding on the genetic diversity of populations . Phil. Trans. R. Soc. Lond. B 358 , 1050–1070 ( doi:10.1098/rstb.2003.1296 ) [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Charlesworth D., Charlesworth B., Morgan M. T.1995 The pattern of neutral molecular variation under the background selection model . Genetics 141 , 1619–1632 [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Crow J. F., Kimura M.1979 Efficiency of truncation selection . Proc. Natl Acad. Sci. USA 76 , 396–399 ( doi:10.1073/pnas.76.1.396 ) [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Eyre-Walker A., Keightley P. D.2009 Estimating the rate of adaptive molecular evolution in the presence of slightly deleterious mutations and population size change . Mol. Biol. Evol. 26 , 2097–2108 ( doi:10.1093/molbev/msp119 ) [ PubMed ] [ Google Scholar ]
- Feldman M. W., Krakauer J.1976 Genetic modification and modifier polymorphisms . In Population genetics and ecology (eds Karlin S., Nevo E.). New York, NY: Academic Press [ Google Scholar ]
- Feldman M. W., Liberman U.1986 An evolutionary reduction principle for genetic modifiers . Proc. Natl Acad. Sci. USA 83 , 4824–4827 [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Feldman M. W., Christiansen F. B., Brooks L. D.1980 Evolution of recombination in a constant environment . Proc. Natl Acad. Sci. USA 77 , 4838–4841 [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Feldman M. W., Otto S. P., Christiansen F. B.1996 Population genetic perspectives on the evolution of recombination . Ann. Rev. Genet. 30 , 261–295 [ PubMed ] [ Google Scholar ]
- Felsenstein J.1974 The evolutionary advantage of recombination . Genetics 78 , 737–756 [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Fisher R. A.1930 The genetical theory of natural selection Oxford, UK: Oxford University Press [ Google Scholar ]
- Funk D. J., Wernegreen J. J., Moran N. A.2001 Intraspecific variation in symbiont genomes: bottlenecks and the aphid—Buchnera association . Genetics 157 , 477–489 [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Gillespie J. H.2000 Genetic drift in an infinite population: the pseudo hitchhiking model . Genetics 155 , 909–919 [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Gillespie J. H.2001 Is the population size of a species relevant to its evolution? Evolution 55 , 2161–2169 [ PubMed ] [ Google Scholar ]
- Haag-Liautard C., Dorris M., Maside X., Macaskill S., Halligan D. L., Houle D., Charlesworth B., Keightley P. D.2007 Direct estimation of per nucleotide and genomic deleterious mutation rates in Drosophila. Nature 445 , 82–85 ( doi:10.1038/nature05388 ) [ PubMed ] [ Google Scholar ]
- Haldane J. B. S.1930 A mathematical theory of natural and artificial selection. VII. Selection intensity as a function of mortality rate . Proc. Camb. Phil. Soc. 27 , 131–136 ( doi:10.1017/S0305004100009427 ) [ Google Scholar ]
- Haldane J. B. S.1957 The cost of natural selection . J. Genet. 55 , 511–524 ( doi:10.1007/BF02984069 ) [ Google Scholar ]
- Halligan D., Keightley P. D.2006 Ubiquitous selective constraints in the Drosophila genome revealed by a genome-wide interspecies comparison . Genome Res. 16 , 875–884 ( doi:10.1101/gr.5022906 ) [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Hamilton W. D.1996 Narrow roads of gene land II Evolution of sex and sexual selection Oxford, UK: W. H. Freeman [ Google Scholar ]
- Hansen T. F.2006 The evolution of genetic architecture . Annu. Rev. Ecol. Evol. Syst. 37 , 123–157 ( doi:10.1146/annurev.ecolsys.37.091305.110224 ) [ Google Scholar ]
- Hellmann I., Ebersberger I., Ptak S. E., Pääbo S., Przeworski M.2005 A neutral explanation of the correlation of diversity with recombination rates in humans . Am. J. Hum. Genet. 72 , 1527–1535 ( doi:10.1086/375657 ) [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Hill W. G., Robertson A.1966 The effect of linkage on limits to artificial selection . Genet. Res 8 , 269–294 ( doi:10.1017/S0016672300010156 ) [ PubMed ] [ Google Scholar ]
- Hudson R. B., Kaplan N. L.1988 The coalescent process in models with selection and recombination . Genetics 120 , 831–840 [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Hudson R. R., Kaplan N. L.1995 Deleterious background selection with recombination . Genetics 141 , 1605–1617 [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Iles M. M., Walters J. R., Cannings C.2003 Selection for recombination in large genomes . Genetics 165 , 2249–2258 [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Johnson T., Barton N. H.2002 The effect of deleterious alleles on adaptation in asexual populations . Genetics 162 , 395–411 [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Kaiser V. B.2009 Molecular evolution under low recombination . PhD thesis, University of Edinburgh, Edinburgh [ PubMed ] [ Google Scholar ]
- Kaiser V., Charlesworth B.2008 The effects of deleterious mutations on evolution in non-recombining genomes . Trends Genet. 25 , 9–12 ( doi:10.1016/j.tig.2008.10.009 ) [ PubMed ] [ Google Scholar ]
- Kaplan N. L., Darden T., Hudson R. B.1988 The coalescent process in models with selection . Genetics 120 , 819–829 [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Keightley P. D., Kacser H.1987 Dominance, pleiotropy and metabolic structure . Genetics 117 , 319–329 [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Keightley P. D., Otto S. P.2006 Interference among deleterious mutations favours sex and recombination in finite populations . Nature 443 , 89–92 ( doi:10.1038/nature05049 ) [ PubMed ] [ Google Scholar ]
- Keightley P. D., Lercher M. J., Eyre-Walker A.2005 Evidence for widespread degradation of gene control regions in hominid genomes . PLoS Biology 3 , e42 ( doi:10.1371/journal.pbio.0030042 ) [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Keightley P. D., Trivedi U., Thomson M., Oliver F., Kumar S., Blaxter M. L.2009 Analysis of the genome sequences of three Drosophila melanogaster spontaneous mutation accumulation lines . Genome Res. 19 , 1195–1201 ( doi:10.1101/gr.091231.109 ) [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Kimura M.1968 Evolutionary rate at the molecular level . Nature 217 , 624–626 ( doi:10.1038/217624a0 ) [ PubMed ] [ Google Scholar ]
- Kimura M., Maruyama T.1966 Mutational load with epistatic gene interactions in fitness . Genetics 54 , 1337–1351 [ PMC free article ] [ PubMed ] [ Google Scholar ]
- King J. L., Jukes T. H.1969 Non-Darwinian evolution . Science 164 , 788–798 ( doi:10.1126/science.164.3881.788 ) [ PubMed ] [ Google Scholar ]
- Kliman R. M., Hey J.1993 Reduced natural selection associated with low recombination in Drosophila melanogaster. Mol. Biol. Evol 10 , 1239–1258 [ PubMed ] [ Google Scholar ]
- Kondrashov A. S.1984 Deleterious mutations as an evolutionary factor. I. The advantage of recombination . Genet. Res. 44 , 199–218 ( doi:10.1017/S0016672300026392 ) [ PubMed ] [ Google Scholar ]
- Kondrashov A. S.1988 Deleterious mutations and the evolution of sexual reproduction . Nature 336 , 435–441 ( doi:10.1038/336435a0 ) [ PubMed ] [ Google Scholar ]
- Kondrashov A. S.1993 Classification of hypotheses on the advantage of amphimixis . J. Hered. 84 , 372–387 [ PubMed ] [ Google Scholar ]
- Kondrashov A. S.1995 Contamination of the genome by very slightly deleterious mutations: why have we not died 100 times over? . J. Theor. Biol. 175 , 583–594 ( doi:10.1006/jtbi.1995.0167 ) [ PubMed ] [ Google Scholar ]
- Lenormand T., Otto S. P.2000 The evolution of recombination in a heterogeneous environment . Genetics 156 , 423–438 [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Lewontin R. C.1974 The genetic basis of evolutionary change New York, NY: Columbia University Press [ Google Scholar ]
- Loewe L.2006 Quantifying the genomic decay paradox due to Muller's ratchet in human mitochondrial DNA . Genet. Res. 87 , 133–159 ( doi:10.1017/S0016672306008123 ) [ PubMed ] [ Google Scholar ]
- Loewe L., Charlesworth B.2006 Inferring the distribution of mutational effects on fitness in Drosophila. Biol. Lett. 2 , 426–430 ( doi:10.1098/rsbl.2006.0481 ) [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Loewe L., Charlesworth B.2007 Background selection in single genes may explain patterns of codon bias . Genetics 175 , 1381–1393 ( doi:10.1534/genetics.106.065557 ) [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Lynch M., Conery J. S.2000 The evolutionary fate and consequences of duplicate genes . Science 290 , 1151–1155 ( doi:10.1126/science.290.5494.1151 ) [ PubMed ] [ Google Scholar ]
- Lynch M., et al.2008 A genome-wide view of the spectrum of spontaneous mutations in yeast . Proc. Natl Acad. Sci. USA 105 , 9272–9277 ( doi:10.1073/pnas.0803466105 ) [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Macpherson J. M., Sella G., Davis J. C., Petrov D. A.2007 Genome-wide spatial correspondence between nonsynonymous divergence and neutral polymorphism reveals extensive adaptation in Drosophila. Genetics 177 , 2083–2099 ( doi:10.1534/genetics.107.080226 ) [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Martin G., Otto S. P., Lenormand T.2006 Selection for recombination in structured populations . Genetics 172 , 593–609 ( doi:10.1534/genetics.104.039982 ) [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Maynard Smith J., Haigh J.1974 The hitch-hiking effect of a favourable gene . Genet. Res. 23 , 23–35 ( doi:10.1017/S0016672300014634 ) [ PubMed ] [ Google Scholar ]
- McVean G. A. T., Charlesworth B.2000 The effects of Hill–Robertson interference between weakly selected sites on patterns of molecular evolution and variation . Genetics 155 , 929–944 [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Merila J., Sheldon B.2000 Lifetime reproductive success and heritability in nature . Am. Nat. 155 , 301–310 ( doi:10.1086/303330 ) [ PubMed ] [ Google Scholar ]
- Muller H. J.1932 Some genetic aspects of sex . Am. Nat. 66 , 118–138 ( doi:10.1086/280418 ) [ Google Scholar ]
- Nachman M. W.2002 Variation in recombination rate across the genome: evidence and implications . Curr. Opin. Genet. Dev. 12 , 657–663 ( doi:10.1016/S0959-437X(02)00358-1 ) [ PubMed ] [ Google Scholar ]
- Nevo E., Beiles A., Ben-Schlomo R.1984 The evolutionary significance of genetic diversity: ecological, demographic and life history correlates . In Measuring selection in natural populations (ed. Mani G. S.), pp. 13–213 Berlin, Germany: Springer [ Google Scholar ]
- Nordborg M., Charlesworth B., Charlesworth D.1996 The effect of recombination on background selection . Genet. Res. 67 , 159–174 ( doi:10.1017/S0016672300033619 ) [ PubMed ] [ Google Scholar ]
- Otto S. P., Feldman M. W.1997 Deleterious mutations, variable epistatic interactions, and the evolution of recombination . Theor. Popul. Biol. 51 , 134–147 ( doi:10.1006/tpbi.1997.1301 ) [ PubMed ] [ Google Scholar ]
- Otto S., Lenormand T.2002 Resolving the paradox of sex and recombination . Nat. Rev. Genet. 3, 252–261 ( doi:10.1038/nrg761 ) [ PubMed ] [ Google Scholar ]
- Peck J. R.1994 A ruby in the rubbish: beneficial mutations and the evolution of sex . Genetics 137 , 597–606 [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Presgraves D. C.2005 Recombination enhances protein adaptation in Drosophila melanogaster. Curr. Biol. 15 , 1651–1656 ( doi:10.1016/j.cub.2005.07.065 ) [ PubMed ] [ Google Scholar ]
- Provine W.1971 The origins of theoretical population genetics Chicago, IL: University of Chicago Press [ Google Scholar ]
- Robertson A.1961 Inbreeding in artificial selection programmes . Genet. Res. 2 , 189–194 ( doi:10.1017/S0016672300000690 ) [ PubMed ] [ Google Scholar ]
- Rouzine I. M., Brunet E., Wilke C. O.2008 The traveling-wave approach to asexual evolution: Muller's ratchet and speed of adaptation . Theor. Popul. Biol. 73 , 24–46 ( doi:10.1016/j.tpb.2007.10.004 ) [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Roze D., Barton N. H.2006 The Hill–Robertson effect and the evolution of recombination . Genetics 173 , 1793–1811 ( doi:10.1534/genetics.106.058586 ) [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Smith N. G. C., Eyre-Walker A.2002 Adaptive protein evolution in Drosophila . Nature 415 , 1022–1024 ( doi:10.1038/4151022a ) [ PubMed ] [ Google Scholar ]
- Spencer C. C. A., Deloukas P., Hunt S., Mullikin J. C., Myers S., Silverman B., Donnelly P., Bentley D., McVean G.2006 The influence of recombination on human genetic diversity . PLoS Genet. 2 , e148 ( doi:10.1371/journal.pgen.0020148 ) [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Stephan W., Langley C. H.1989 Molecular genetic variation in the centromeric region of the X chromosome in three Drosophila ananassae populations I Contrasts between the vermilion and forked loci . Genetics 121 , 89–99 [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Stephan W., Wiehe T. H., Lenz M.1992 The effect of strongly selected substitutions on neutral polymorphism: analytical results based on diffusion theory . Theor. Popul. Biol. 41 , 237–254 ( doi:10.1016/0040-5809(92)90045-U ) [ Google Scholar ]
- Sved J. A.1968 Possible rates of gene substitution in evolution . Am. Nat. 102 , 283–293 ( doi:10.1086/282542 ) [ Google Scholar ]
- Sved J. A., Reed T. E., Bodmer W. F.1967 The number of balanced polymorphisms that can be maintained by natural selection . Genetics 55 , 469–481 [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Szathmary E.1993 Do deletion mutations act synergistically? Metabolic control theory provides a partial answer . Genetics 133 , 127–132 [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Wakeley J.2008 Coalescent theory: an introduction Englewood, CO: Roberts and Company [ Google Scholar ]
- Waxman D., Peck J. R.1999 Sex and adaptation in a changing environment . Genetics 153 , 1041–1053 [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Weismann A.1889 The significance of sexual reproduction in the theory of natural selection . In Essays upon heredity and kindred biological problems (eds Poulton E. B., Schönland S., Shipley A. E.). Oxford, UK: Clarendon Press [ Google Scholar ]
- Williams G. C.1975 Sex and evolution Princeton, NJ: Princeton University Press [ Google Scholar ]

Gene flow between species influences evolution in Darwin’s finches
Princeton ecologists Peter and Rosemary Grant led a team of researchers to discover how genetics and hybridization affected the beak shape of finches on the Galápagos Islands, such as this medium ground finch with its characteristic blunt beak. This particular specimen was banded by the husband-and-wife team during their field studies on Daphne Major.
Despite the traditional view that species do not exchange genes by hybridization, a new study led by Princeton ecologists Peter and Rosemary Grant show that gene flow between closely related species is more common than previously thought.
A team of scientists from Princeton University and Uppsala University detail their findings of how gene flow between two species of Darwin’s finches has affected their beak morphology in the May 4 issue of the journal Nature Ecology and Evolution.
Darwin’s finches on the Galápagos Islands are an example of a rapid adaptive radiation in which 18 species have evolved from a common ancestral species within a period of 1 to 2 million years. Some of these species have only been separated for a few hundred thousand years or less.
Rosemary and Peter Grant of Princeton University, co-authors of the new study, studied populations of Darwin’s finches on the small island of Daphne Major for 40 consecutive years and observed occasional hybridization between two distinct species, the common cactus finch and the medium ground finch. The cactus finch (Geospiza scandens) is slightly larger than the medium ground finch (G. fortis), has a more pointed beak and is specialized to feed on cactus. The medium ground finch has a blunter beak and is specialized to feed on seeds.

Schematic figure showing the outcome of hybridization between male cactus finches and female ground finches. Rosemary and Peter Grant have studied these birds on the small island of Daphne Major for more than 40 years. The common cactus finch has a pointed beak adapted to feed on cactus, whereas the medium ground finch has a blunt beak adapted to crush seeds. Hybrid females successfully mate with male cactus finch males, whereas the hybrid males do not successfully compete for high quality territory and mates. Because these hybrid females receive their single Z chromosome from their cactus finch father there is no gene flow on Z chromosomes between species through these hybrid females.
“Over the years, we observed occasional hybridization between these two species and noticed a convergence in beak shape,” said the husband-and-wife team, who have been research partners for decades. Peter Grant is the emeritus Class of 1877 Professor of Zoology and an emeritus professor of ecology and evolutionary biology, and Rosemary Grant is an emeritus senior research biologist.
“In particular, the beak of the common cactus finch became blunter and more similar to the beak of the medium ground finch,” continued the Grants. “We wondered whether this evolutionary change could be explained by gene flow between the two species.”
“We have now addressed this question by sequencing groups of the two species from different time periods and with different beak morphology,” said Sangeet Lamichhaney, one of the shared first authors and an associate professor at Kent State University. “We provide evidence of a substantial gene flow, in particular from the medium ground finch to the common cactus finch.”
“A surprising finding was that the observed gene flow was substantial on most autosomal chromosomes but negligible on the Z chromosome, one of the sex chromosomes,” said Fan Han, a graduate student at Uppsala University, who analysed these data as part of her Ph.D. thesis. “In birds, the sex chromosomes are ZZ in males and ZW in females, in contrast to mammals where males are XY and females are XX.”
“This interesting result is in fact in excellent agreement with our field observation from the Galápagos,” said the Grants. “We noticed that most of the hybrids had a common cactus finch father and a medium ground finch mother. Furthermore, the hybrid females successfully bred with common cactus finch males and thereby transferred genes from the medium ground finch to the common cactus finch population. In contrast, male hybrids were smaller than common cactus finch males and could not compete successfully for high-quality territories and mates.”

Common cactus finch with its pointed beak feeding on the Opuntia cactus.
This mating pattern is explained by the fact that Darwin’s finches imprint on the song of their fathers, so sons sing a song similar to their father’s song and daughters prefer to mate with males that sing like their fathers. Furthermore, hybrid females receive their Z chromosome from their cactus finch father and their W chromosome from their ground finch mother. This explain why genes on the Z chromosome cannot flow from the medium ground finch to the cactus finch via these hybrid females, whereas genes in other parts of the genome can, because parents of the hybrid contribute equally.
“Our data show that the fitness of the hybrids between the two species is highly dependent on environmental conditions which affect food abundance — that is, to what extent hybrids, with their combination of gene variants from both species, can successfully compete for food and territory,” said Leif Andersson of Uppsala University and Texas A&M University.
He continued: “The long-term outcome of the ongoing hybridization between the two species will depend on environmental factors as well as competition. One scenario is that the two species will merge into a single species combining gene variants from the two species, but perhaps a more likely scenario is that they will continue to behave as two species and either continue to exchange genes occasionally or develop reproductive isolation if the hybrids at some point show reduced fitness compared with purebred progeny. The study contributes to our understanding of how biodiversity evolves.”
“ Female-biased gene flow between two species of Darwin’s finches ,” by Sangeet Lamichhaney, Fan Han, Matthew T. Webster, B. Rosemary Grant, Peter R. Grant and Leif Andersson, appeared in the May 4 issue of Nature Ecology & Evolution (DOI: 10.1038/s41559-020-1183-9 ). The research was supported by the Galápagos National Parks Service, the Charles Darwin Foundation, the National Science Foundation, the Knut and Alice Wallenberg Foundation and the Swedish Research Council.
Related Stories

Study of Darwin's finches reveals that new species can develop in as little as two generations .
The arrival 36 years ago of a strange bird to a remote island in the Galápagos archipelago has provided direct genetic evidence of a novel way in which new species arise.
A gene that shaped the evolution of Darwin's finches .
Researchers from Princeton University and Uppsala University in Sweden have identified a gene in the Galápagos finches studied by English naturalist Charles Darwin that influences beak shape and that played a role in the birds' evolution from a common ancestor more than 1 million years ago. The study illustrates the genetic foundation of evolution, including how genes can flow from one species to another, and how different versions of a gene within a species can contribute to the formation of entirely new species.
Gene behind 'evolution in action' in Darwin's finches identified .
Scientists from Princeton University and Uppsala University have identified a specific gene that within a year helped spur a permanent physical change in a finch species in response to a drought-induced food shortage. The findings provide a genetic basis for natural selection that, when combined with observational data, could serve as a comprehensive model of evolution.
Noted Princeton husband-and-wife team wins Kyoto Prize .
Princeton University's Peter and Rosemary Grant, whose legendary explorations on the bleak Galapagos island of Daphne Major over nearly four decades have produced an array of dazzling insights into evolutionary theory, have been named recipients of the Kyoto Prize.
Lecture honors Kyoto Prize-winning Grants .
Princeton scientists Peter and Rosemary Grant, winners of this year's Kyoto Prize for their pioneering work in evolutionary biology, will be honored with a lecture by noted researcher Jonathan Losos at 6 p.m. Friday, Dec. 4, in 10 Guyot Hall.

Peter and Rosemary Grant receive Royal Medal in Biology .
Peter Grant, the Class of 1877 Professor of Zoology, Emeritus, and professor of ecology and evolutionary biology, emeritus, and B. Rosemary Grant, senior biologist, ecology and evolutionary biology, have been named recipients of the Royal Medal in Biology.
Following in Darwin’s footprints: Hau unlocks secrets of tropical birds through field study on the Galápagos .
The Galápagos Islands hold a unique place in the history of science. It was here, in the 1830s, that Charles Darwin gathered the clues that led to his theory of evolution. It is here, today, that Princeton’s Michaela Hau continues Darwin’s intrepid scientific tradition. Her studies of tropical birds may shed light on the mysteries of human behavior and could lead to better models for ecosystem conservation.
HYPOTHESIS AND THEORY article
Nasal turbinate lymphatic obstruction: a proposed new paradigm in the etiology of essential hypertension.

- 1 Department of Radiology, UT-Health San Antonio, San Antonio, TX, United States
- 2 Department of Pathology, Methodist Hospital, San Antonio, TX, United States
Hypertension affects an estimated 1.3 billion people worldwide and is considered the number one contributor to mortality via stroke, heart failure, renal failure, and dementia. Although the physiologic mechanisms leading to the development of essential hypertension are poorly understood, the regulation of cerebral perfusion has been proposed as a primary cause. This article proposes a novel etiology for essential hypertension. Our hypothesis developed from a review of nuclear medicine scans, where the authors observed a significantly abnormal increase in nasal turbinate vasodilation in hypertensive patients using quantitative region of interest analysis. The authors propose that nasal turbinate vasodilation and resultant blood pooling obstruct the flow of cerebrospinal fluid passing through nasal turbinate lymphatics, thereby increasing intracranial pressure. The authors discuss the glymphatic/lymphatic clearance system which is impaired with age, and at which time hypertension also develops. The increased intracranial pressure leads to compensatory hypertension via Cushing’s mechanism, i.e., the selfish brain hypothesis. The nasal turbinate vasodilation, due to increased parasympathetic activity, occurs simultaneously along with the well-established increased sympathetic activity of the cardiovascular system. The increased parasympathetic activity is likely due to an autonomic imbalance secondary to the increase in worldwide consumption of processed food. This hypothesis explains the rapid worldwide rise in essential hypertension in the last 50 years and offers a novel mechanism and a new paradigm for the etiology of essential hypertension. This new paradigm offers compelling evidence for the modulation of parasympathetic nervous system activity as a novel treatment strategy, specifically targeting nasal turbinate regulation, to treat diseases such as hypertension, idiopathic intracranial hypertension, and degenerative brain diseases. The proposed mechanism of essential hypertension presented in this paper is a working hypothesis and confirmatory studies will be needed.
Hypertension affects an estimated 1.3 billion people worldwide and is considered the number one contributor to mortality via stroke, heart failure, renal failure, and dementia. Although the physiologic mechanisms leading to the development of essential hypertension are poorly understood, the regulation of cerebral perfusion has been proposed as a primary cause.
Our hypothesis regarding the etiology of hypertension developed from a retrospective review of 200 nuclear medicine scans, where the authors observed a significant increase in nasal turbinate vasodilation in hypertensive patients. The authors propose that the nasal turbinate vasodilation and subsequent increased blood pooling obstruct the flow of cerebrospinal fluid passing through nasal turbinate lymphatics, thereby increasing intracranial pressure. The increased intracranial pressure leads to compensatory arterial hypertension via Cushing’s mechanism. This hypothesis offers a novel mechanism and a new paradigm for the etiology of essential hypertension related to nasal turbinate obstruction of brain lymphatics and suggests possible new treatments for hypertension and degenerative brain diseases. Treating hypertension by methods that focus on nasal turbinate obstruction and/or increasing cerebrospinal fluid lymphatic flow through the nasal turbinates may offer a therapeutic benefit not only to hypertensive patients but to patients with neurodegenerative pathologies as well.
1 Introduction
Essential hypertension, also known as primary hypertension, affects an estimated 1.3 billion people worldwide and is considered the number one contributor to mortality via stroke, heart failure, renal failure, and dementia. It is the largest single contributor to global mortality ( 1 ). Each year approximately 10 million people worldwide die of hypertension-related disease. The prevalence of essential hypertension is increasing. Between 1990 and 2019, the number of people aged 30–79 years with hypertension doubled from 331 million women and 317 million men in 1990 to 626 million women and 652 million men in 2019 ( 2 , 3 ). The number of individuals with essential hypertension has steadily increased over the past few decades, likely associated with the large increase in overweight and obese individuals in the world ( 4 , 5 ). In the United States, the lifetime risk of hypertension surpasses 80% ( 6 ). Currently, half of all adults in the United States have hypertension, and the disease is responsible for the highest percentage of all doctor visits ( 6 ).
Today nearly 70 percent of what individuals eat in the United States is ultra-processed food. These foodstuffs include packaged chips, energy drinks, and ready-to-heat-and-eat meals. They are thought to be an important driver of the obesity epidemic, in part because they seem to make us eat more ( 7 ). This obesity epidemic occurring in the United States has also been noted in other developed and developing countries throughout the world.
Changes in dietary patterns in China, with increased consumption of refined grains and highly processed, high-sugar, and high-fat foods, continue to increase while physical activity levels in all major domains have decreased ( 5 ). In China, the number of processed foods available was four times higher in 2013 than in 1999 for a 22.4% annual growth over the 15 years. Over half of the packaged foods sold in China’s markets are processed foods. Overweight, obesity, hypertension, and metabolic syndrome in the Chinese population have become serious public health problems. In 2015, China had the highest number of overweight and obese children globally ( 5 ). The increased rate of obesity and hypertension in China likely explains the fact that stroke is now the number one cause of death in that country ( 8 ).
Essential hypertension or primary hypertension is not equally distributed in populations worldwide. In the United States, essential hypertension accelerates more rapidly in non-Hispanic Black individuals (NHB) than in non-Hispanic White individuals (NHW) and is often more severe with higher mortality ( 9 ). In 2020, age-adjusted hypertension-related NHB adult death rates were approximately twice that of NHW adults (325.3 thousand for NHB men compared with 175.7 thousand for NHW men and 216.1 for NHB women compared with 127.9 for NHW women) ( 9 ).
In current medical practice, lifestyle changes are often mentioned as a first line of therapy for patients with hypertension. Alterations or modifications in diet, such as the Dietary Approaches to Stop Hypertension (DASH) ( 10 ) are encouraged, as are increased exercise, and restriction of sodium intake. Lowering salt intake moderately reduces blood pressure. An updated systematic review of studies where sodium intake was reduced from 2,200 mg/day to 500 mg/day for 1 week found that the median within-individual change in mean arterial blood pressure between high and low sodium diets was 4 mmHg ( 11 ). A newer, more invasive therapeutic technique to control high blood pressure involves renal nerve ablation which reduces sympathetic nervous system activity in the kidney ( 12 ).
Today, the most commonly prescribed antihypertensive drugs according to the latest guidelines are combination drug therapies that block the renin-angiotensin system and increase sodium excretion ( 6 ). With the rise of renin-angiotensin-targeted drugs, therapies that specifically target only the sympathetic nervous system have significantly decreased in use, even though one of the most verified findings in essential hypertension is that increased sympathetic nervous system activity is associated with the onset of hypertension ( 12 – 16 ). The etiology of this increased sympathetic activity, however, remains controversial as discussed in a recent review of autonomic dysfunction in essential hypertension ( 13 ).
Although many different theories about the cause of essential hypertension have been proposed, including excessive salt intake, renal mechanisms, and stress, for most adults there is no clearly identifiable cause with many investigators ascribing the mechanisms of hypertension to multiple factors including interactions between diet and lifestyle, an individual’s gut microbiome ( 17 ), neuroimmune modulation ( 18 ), and genetic ( 17 ) and epigenetic factors ( 19 ).
It is well known that hypertension is found in families and that there is a hereditary predisposition to developing hypertension with over 100 single nucleotide polymorphisms associated with the disease ( 17 ). Secondary hypertension , as differentiated from primary or essential hypertension, has a higher prevalence in children (50% of cases) and young adults less the 30 years of age. Hormonal and primary kidney disease are the main causes of secondary hypertension. Genetic predisposition interacting with environmental influences is a significant contributor to the development of hypertension with the clearest genetic linkages being evident in endocrine hypertension, a form of secondary hypertension ( 20 ). Endocrine hypertension has well-defined phenotypes that have allowed patient stratification into homogeneous cohorts. These cohorts can be linked to different genetic variants which have important implications concerning patient therapy ( 21 ). Primary aldosteronism is the most frequent form of endocrine hypertension accounting for 5–10% of all hypertensive patients. Several different genetic defects have been linked to primary aldosteronism including autosomal dominant forms and somatic mutations ( 20 ).
Although genetic predisposition plays a role in essential primary hypertension , the genetic linkages are more complex. In addition, there are significant environmental influences making identification of specific genetic linkages less clear. Although the genetic linkages are less clear, genetic heritability is estimated to account for 40% of blood pressure variance in essential hypertension while environmental influences such as dietary and lifestyle habits can explain the majority of the remaining genetic variance ( 21 ). A promising application in the field of hypertension is the use of genetic testing to personalize medical therapy by predicting which anti-hypertensive drugs are most likely to have the greatest effect or cause adverse reactions in an individual patient ( 22 ).
Most researchers continue to state that the primary cause of non-endocrine essential hypertension is not well understood ( 14 , 23 ). This is likely related to the fact that essential hypertension is a multifactorial disease that is considered to be genetically complex with significant interactions with diet and epigenetic factors.
A lack of understanding of the mechanism of essential hypertension contributes to the fact that an estimated 10–30% of patients have resistant hypertension defined as blood pressure that remains above guideline-directed targets despite the use of three anti-hypertensives (including a diuretic) at optimal or maximally tolerated doses ( 24 ). Other studies report that the global control rate of blood pressure among people with hypertension was approximately 20% in 2019 ( 2 ). A possible explanation for this overall lack of blood pressure control is that the underlying basic pathophysiology leading to the development of essential hypertension is not being addressed. Hence, there is a need to develop new paradigms for understanding essential hypertension with the potential to develop new approaches to therapy.
The objective of this paper is to focus on areas not previously considered as the pathogenesis of hypertension. The authors hypothesize that there is an increase in parasympathetic activity in the nasal turbinates that relates to hypertension by causing obstruction of nasal lymphatic drainage, thereby increasing intracranial pressure. The increased intracranial pressure leads to compensatory hypertension via Cushing’s mechanism, also known as the selfish brain. This increased parasympathetic activity of the nasal turbinates occurs simultaneously with the well-established increase in sympathetic nervous activity of the cardiovascular system in hypertension. The increased nasal turbinate vasodilation has been previously described in patients with essential hypertension and other metabolic syndrome features in a recent article by the authors ( 25 ). This increased parasympathetic activity results not only in nasal turbinate vasodilation, but also in increased gastrointestinal motility, as observed in hypertensive patients and other patients with metabolic syndrome ( 26 – 29 ).
2 Regulation of cerebral perfusion
A less frequently discussed proposed cause of essential hypertension is related to homeostatic processes for the regulation of cerebral perfusion ( 14 , 30 ). Physiologic processes that impair blood flow to the brain have the potential to lead to increased sympathetic activity and elevated systemic blood pressure to maintain normal blood flow to the brain. The theory has been proposed as the “selfish brain hypothesis of essential hypertension” or “Cushing’s mechanism” ( 14 , 30 ). Decreased blood flow to the brain and subsequent development of hypertension via the Cushing mechanism has been previously reported to be associated with the narrowing of the vessels supplying the brain ( 31 ).
This paper proposes another potential mechanism for the decreased blood flow to the brain that leads to systemic hypertension. It focuses on clinical findings by the authors of increased nasal turbinate vasodilation and resultant nasal blood pooling that causes a restriction of lymphatic flow, or drainage, of cerebrospinal fluid (CSF) from the brain ( 25 ). The obstruction of drainage leads to increased intracranial pressure, resulting in increased systemic blood pressure via Cushing’s mechanism ( 14 , 30 ).
The objective of this paper is to review the literature regarding the above-described physiological mechanisms of hypertension—noting the potential influence of the parasympathetic nervous system on increased intracranial pressure—and to propose a novel etiology for this increasingly prevalent disease.
3 Increased intracranial pressure and Cushing’s mechanism
3.1 hypertension and increased intracranial pressure.
Increased intracranial pressure is present in patients with essential hypertension ( 32 ). In 2023, da Costa et al. ( 32 ) studied 391 consecutive patients with long-term essential hypertension in an attempt to evaluate intracranial pressure waveforms using a non-invasive device, brain4care. Their study revealed 77.7% of the patients had abnormal measurements of intracranial pressure. The da Costa et al. ( 32 ) article was the first to evaluate intracranial pressure behavior in patients with essential hypertension. In addition to their findings, the authors commented that very little is known on the subject of intracranial pressure in patients with hypertension and that they were hoping to “shed some light on the dark side of human history.”
This increased intracranial pressure in hypertensive patients is consistent with the authors’ research showing increased nasal turbinate vasodilation in these same patients. We hypothesize that inappropriately increased nasal turbinate vasodilation with blood pooling in the nasal turbinates is obstructing the normal lymphatic drainage through the nasal turbinates, resulting in increased intracranial pressure.
The authors can find no instances in the medical literature in which invasive lumbar puncture CSF pressure measurements have been performed to study intracranial pressure in patients with essential hypertension. Performing this type of study would be fairly extensive since it is likely the elevations of CSF pressure in many patients would be mild, although significant, in relation to its potential association with essential hypertension.
3.2 Cushing’s mechanism
The regulation of cerebral perfusion has been proposed as a cause of essential hypertension with cerebral perfusion pressure being preserved by an increase in systemic blood pressure secondary to increased sympathetic activity ( 14 , 33 – 36 ). As early as 1901, Dr. Harvey Cushing proposed the idea of a “Cushing reflex” which he described as a physiological nervous system response to acute elevations of intracranial pressure (ICP) ( 37 , 38 ). The response consisted of a triad of signs which included widened pulse pressure (increasing systolic, decreasing diastolic), bradycardia, and irregular respirations. He believed that the dramatic increase in blood pressure was a reflex to brainstem ischemia seen in patients with increasing ICP from causes such as intracranial hemorrhage, a mass effect from a tumor, cerebral edema, and other causes. In these studies, Cushing showed that a temporary reduction in cerebral blood flow secondary to increased ICP was associated with a compensatory increase in systemic blood pressure in animals ( 38 ). This increase in systemic blood pressure was part of a regulatory process to maintain normal cerebral blood flow.
The human brain is in a tight space, limited by the rigid skull, which makes for a unique situation as it relates to blood and lymphatic flow rates and the strict requirement of the brain to maintain adequate cerebral blood perfusion. Cerebral perfusion pressure is the pressure that pushes the blood through the cerebrovascular network. Cerebral perfusion pressure is a clinical surrogate for the adequacy of cerebral blood perfusion. Cerebral perfusion pressure (CPP) is equal to the mean arterial pressure (MAP) minus the intracranial pressure (ICP) in the following equation ( 39 ).
MAP can be estimated as the systolic blood pressure (SBP) plus two times the diastolic blood pressure (DBP) divided by 3.
As intracranial pressure increases, the cerebral perfusion pressure (CPP) decreases unless there is a compensatory increase in mean arterial blood pressure ( 14 , 33 , 34 ). With increased ICP, MAP must also increase to maintain adequate blood flow in the brain or CPP. This relationship between ICP and CPP was originally shown by Cushing ( 37 ). The authors believe nasal turbinate vasodilatation and subsequent blood pooling obstruct the normal drainage of cerebrospinal fluid from the brain. This obstruction results in increased intracranial pressure (ICP), requiring a compensatory increase in mean arterial pressure (MAP) to maintain cerebral perfusion pressure (CPP). How and where the brain senses its blood flow and is then able to maintain a normal blood flow by increasing systemic blood pressure via the increased sympathetic activity of the heart and vasculature is still a matter of debate.
3.3 Cerebral blood flow resistance and hypertension
As early as 1948, Kety et al. ( 34 ) reported that patients with essential hypertension had increased cerebral vascular resistance. Their article states that “there is at least some evidence to favor the hypothesis that in essential hypertension there may be a primary cerebrovascular constriction accompanied by a secondary and compensatory hypertension which maintains a normal cerebral blood flow.”
Other researchers ( 40 , 41 ) confirmed the findings of Kety et al. ( 34 ) that cerebrovascular resistance is increased in hypertension and that increased cerebral vascular resistance is the best predictor of the future development of hypertension.
Increased cerebrovascular resistance and increased intracranial pressure have been linked to increased sympathetic activity. In 2018, Schmidt et al. ( 42 ) showed that small increases in intracranial pressure would induce a significant increase in sympathetic activity in mice and in humans. In their study in human patients, a 7 mmHg rise in intracranial pressure increased sympathetic muscle activity by 17% as measured by microneurography. This increased sympathetic activity was associated with an elevation in blood pressure.
4 Hypertension: a mechanism for self-protection of the brain
Various authors have proposed that hypertension may provide self-protection for the brain by maintaining normal cerebral blood flow as suggested in the selfish brain hypothesis ( 14 , 43 ).
In 1990, Dickinson ( 33 ) wrote an article reappraising the importance of the Cushing reflex for blood pressure stabilization. In this article, he stated that a restriction of blood flow to the brain can produce sustained hypertension. Dickinson also stressed the fact that the Cushing response begins when intracranial pressure begins to rise and is still within the physiological range. He described the Cushing reflex as the most powerful neural blood pressure stabilizing system involving self-protection of the brain.
Paton et al. ( 30 ) revisited the idea of self-protection of the brain in 2009, stating specifically that brainstem hypoperfusion could cause the onset of sympathetic hyperactivity and hypertension. They called this the “Cushing’s mechanism,” which was later termed “the selfish brain hypothesis” in an article by Hart ( 14 ). Warnert et al. ( 43 ) wrote an article that asked the question “Is high blood pressure self-protection for the brain?.” In support of the increase in brain blood flow resistance as a cause of essential hypertension, Hart suggested that congenital vertebral artery hypoplasia is a risk factor for essential hypertension ( 14 ). Further studies by her group found that vertebral artery hypoplasia plus an incomplete circle of Willis was associated with lower cerebral blood flow in young adults with hypertension ( p = 0.0172) ( 44 ). This anatomical variant was predictive of hypertension in young adults.
Although this work provides support for the selfish brain hypothesis for subjects with vertebral artery hypoplasia, it would not appear to explain the common and worldwide occurrence of essential hypertension and its near doubling in the number of affected individuals over the last 20 years ( 2 ). Instead, the rapidly increasing incidence of hypertension is more consistent with environmental changes likely related to decreased physical activity and diet.
5 The autonomic nervous system
5.1 the parasympathetic nervous system vs. the sympathetic nervous system.
The autonomic nervous system consists of the sympathetic and parasympathetic nervous systems. The sympathetic nervous system controls “flight-or-fight” responses. It prepares the body for strenuous physical activity by increasing the heart rate, elevating blood pressure, heightening awareness, and elevating the respiratory rate. The parasympathetic nervous system carries signals to relax those systems and bring about a state of calm in the body. Parasympathetic responses include an increase of digestive enzymes and more rapid gastric emptying ( 45 ), dilation of nasal turbinate blood vessels ( 46 ), and decreased heart rate ( 47 ).
5.2 The paradox of increased sympathetic activity and concurrent increased parasympathetic activity
Perhaps the most verified and agreed upon finding in essential hypertension is increased sympathetic nerve activity ( 14 , 36 , 48 – 50 ). Sympathetic nerve activity, measured by direct microneurography, was found to be increased in hypertension, providing evidence of the involvement of increased sympathetic activity in the development of essential hypertension. Wallin et al. ( 51 ) were the first to measure sympathetic nerve activity of the peritoneal nerve and showed that sympathetic nerve activity was increased in hypertensive patients as compared to normotensive patients. Subsequently, the increased sympathetic activity of the cardiovascular system has been confirmed by many investigators ( 14 , 36 , 48 , 52 , 53 ).
Increased sympathetic activity clearly affects the cardiovascular system . How increased sympathetic activity affects other organ systems is less well understood, although it has generally been assumed that other organ systems in patients with hypertension experience increased sympathetic activity. As support for the increased sympathetic activity in many organ systems, one article cites a decrease in salivary flow associated with hypertension ( 54 ) and suggests that there is a down-regulation of parasympathetic activity in all organ systems. However, our group reported nuclear imaging-based findings in hypertensive patients that are consistent with increased parasympathetic activity in several non-cardiovascular systems ( 25 , 26 ). Increased vasodilation of the nasal turbinates and parotid glands in hypertensive patients, recently reported by our group ( 25 ), is consistent with increased parasympathetic activity affecting the vasculature of the nasal turbinates, as increased sympathetic activity is well known to be associated with nasal turbinate and parotid vascular constriction . Abnormally rapid gastric emptying in hypertensive patients, as previously reported by our group, is consistent with increased parasympathetic activity ( 26 ), as increased sympathetic activity would have the opposite effect, and inhibit gastrointestinal motility. In our study, following ingestion of a liquid carbohydrate meal, hypertensive patients had an average of 41% more rapid gastric emptying compared to non-hypertensive patients ( p = 0.02), and the rate of gastric emptying correlated significantly with the postprandial glucose level at 30 min (Spearman rank correlation coefficient rs = 0.64, p = 0.0428). Our group has also reported that abnormally rapid gastric emptying occurs in spontaneously hypertensive rats (SHR) ( 55 ). This rapid gastric emptying observed in humans and in rat models with hypertension is consistent with increased parasympathetic activity of the gastrointestinal system.
As far as the authors know, this paradox of increased sympathetic activity in one region (cardiovascular) while there is simultaneously increased parasympathetic activity in another region has not been previously described ( 25 ). This paradox is important for the pathology we will be discussing related to the potential lymphatic obstruction from the brain.
6 New glucose set points
The upregulation, or increase in parasympathetic activity, affecting both the gastrointestinal system and nasal turbinate vasodilation that we have observed in our clinical patients, may be related to glucose homeostasis. The authors hypothesize that the increased parasympathetic activity observed in hypertensive patients is due to a resetting of the body’s glucose level. The elevated blood glucose level, or elevated glucose set point, causes a triggering mechanism for an increase in parasympathetically controlled gastric emptying as a means to sustain the elevated glucose levels. Prior studies have shown that an increased gastric emptying rate is an important mechanism for maintaining blood glucose levels ( 56 – 58 ). The increased rate of gastric emptying occurs due to signaling from the hypothalamus via the vagus nerve ( 59 , 60 ). With a higher glucose set point level, food empties more rapidly from the stomach for absorption into the intestine to elevate and maintain blood glucose levels.
Although the mechanism by which glucose set points become elevated is not clearly understood, the authors hypothesize they become gradually elevated due to the continual increased intake of processed foods. The modern diet, consisting of ultra-processed products, sucrose, and refined grains combined with reduced consumption of fiber, fruits, and vegetables, results in elevated postprandial glucose levels and an upward resetting of the glucose set point. This elevated glucose set point hypothesis is consistent with the significant increase in obesity which has nearly tripled in prevalence since 1960, and the nearly doubling of the number of patients with hypertension over the last 20 years ( 2 ).
Our group was the first to report that a gastrointestinal hormone, cholecystokinin (CCK-8), which delays the rate of gastric emptying in patients, had the potential to treat diabetes by lowering postprandial glucose levels ( 61 ). We reported that many patients with type 2 diabetes have abnormally accelerated gastric emptying and that infusion of CCK-8 significantly reduced the rate of gastric emptying, which lowered postprandial glucose levels ( 61 ). The clinically approved intestinal hormone, glucagon-like peptide 1 (GLP-1), has been widely successful in the treatment of diabetes and obesity. GLP-1 also delays gastric emptying and decreases postprandial glucose levels. Based on the results of our previous studies, GLP-1 would therefore lead to a lowering of the glucose set point. This hypothesis is consistent with the recent findings that GLP-1 agents have been shown to decrease the incidence of cardiovascular disease and stroke in patients with obesity and without diabetes ( 62 ). Importantly, GLP-1 drugs have also been shown to result in a modest (5–7 mmHg) lowering of blood pressure that is greater than would be expected from weight loss alone ( 63 – 65 ).
7 Nasal turbinate vasodilation and blood pooling
If increased intracranial pressure is indeed a frequent etiology for hypertension, how is it possible, much less probable, that millions of individuals with hypertension have preexisting increased intracranial pressure? The authors believe that increased parasympathetic activity leads to vasodilation of the erectile tissue of the nasal turbinates. These nasal turbinates contain important lymphatic vessels that carry spinal fluid moving through the cribriform plate along the olfactory nerves. We hypothesize that this nasal turbinate vasodilation and blood pooling obstruct the lymphatic cerebrospinal fluid (CSF) drainage leading to increased intracranial pressure.
In a retrospective study of 200 patients referred for a routine bone scan, the authors observed that hypertensive patients have significant nasal blood pooling, i.e., increased nasal turbinate vasodilation, as compared to patients without hypertension ( 25 ). This increased nasal vasodilation in patients with hypertension is illustrated in Figure 1 . The methodology used for obtaining the nuclear scan and the whole-body blood pool imaging is described in the following section.

Figure 1 . Weight-matched, non-hypertensive (A) vs. hypertensive patient (B) , both with normal BMIs.
8 Scintigraphic imaging
Scintigraphic imaging of the nasal blood activity in comparison with the cardiac blood activity was determined during the 7-min interval immediately following injection of a bone avid radiopharmaceutical, technetium-99 m methylene diphosphonate ( 99m Tc-MDP), when the radioactivity was in the blood, and before it had time to begin accumulating in the bone. The same scintigraphic imaging technique was used for all 200 patients in this retrospective analysis of whole-body blood pool scanning. Each scan was obtained beginning 2–3 min after injection of the bone avid radiopharmaceutical and took a total of 6–7 min to scan from head to feet. The early images of the bone avid radiopharmaceutical, within the first few minutes after injection, are considered to be markers of the patient’s blood pool, as the radiopharmaceutical requires approximately 3 h for bone deposition and clearance of activity from the soft tissues. Images were obtained with a dual-headed gamma camera (GE Infinia Hawkeye 4, Boston, MA) using low-energy, high-resolution collimators with an energy window set at 140 keV and with a 20% window moving at a rate of 36 cm/min ( 25 ). With scintigraphic imaging, it is possible to determine the distribution and activity of blood in the nasal region as compared to the cardiac region.
9 Measurement of nose/heart ratios
Nose/heart ratios were determined by placing a square region of interest box over the area of the nose on the nuclear scan. The activity in the maximum pixel was determined in each box, and a ratio of the maximum pixel in the nose was divided by the maximum pixel in the heart. Using the maximum pixel activity is very similar in technique to analyzing the maximum standard uptake value (MaxSUV) as determined in PET imaging for monitoring cancer metabolism. The use of a box and maximum pixel activity decreases the subjectivity incurred with drawing an outline around the whole organ. In our retrospective study of 200 patients, those patients with hypertension had an average nose-to-heart max ratio of 0.93 versus 0.85 in non-hypertensive patients ( p = 0.0123 using the Wilcoxon rank-sum test) ( 25 ). Figure 1 demonstrates a normal-weight non-hypertensive control subject (A) compared to a normal-weight hypertensive patient with increased nasal pooling (B).
10 Nasal blood pooling observations on nuclear medicine scan
10.1 increased nasal blood pooling in a normal weight-matched, hypertensive patient vs. a non-hypertensive patient, 10.1.1 increased nasal blood pooling in weight-matched, obese, hypertensive vs. non-hypertensive patients.
Figure 2 illustrates the nuclear medicine scan of weight-matched patients, both with an elevated body mass index (BMI). Patient A is a normal control and Patient B has hypertension and hyperlipidemia, but not diabetes or sleep apnea. There is increased nasal blood pooling in the overweight hypertensive patient compared to the overweight non-hypertensive control subject.

Figure 2 . Weight-matched, non-hypertensive (A) vs. hypertensive patient (B) , both with elevated BMIs.
Both non-hypertensive patients in Figures 1 , 2 have very minimal blood activity in their nasal turbinates while both patients with hypertension have very significant activity in the nasal turbinate region. These whole-body blood pool imaging studies have provided insights to the investigators which have led to their proposal of a working hypothesis described in this paper regarding a new causation paradigm for essential hypertension. Confirmation of these findings will be important. Potential methods to confirm these findings will be addressed in section 19.
10.2 Increased blood pooling in erectile tissue of the nasal turbinates
Studies of computed tomography (CT) ( 66 ) and magnetic resonance imaging (MRI) ( 67 ) have shown that the erectile tissue in the nose is located in (1) the whole of the inferior turbinate (anterior, middle, and posterior), (2) the middle turbinate (more prominent at the middle and posterior turbinate), and (3) the anterior portion of the nasal septum.
The rapidity in which these turbinates can dilate and contract has led two different investigators, Cole et al. ( 66 ) and Ng et al. ( 67 ), to conclude that nasal turbinate dilation is due to an increase in blood volume in the nasal turbinates and not due to an increase in edema or interstitial fluid. This purported increase in nasal blood pool volume is consistent with our findings of high nasal blood activity in the turbinate region observed on nuclear imaging.
The vasodilation associated with increased nasal turbinate parasympathetic activity is the opposite of the vasoconstrictive effect of sympathetic activity on the nasal turbinates and the well-known vasoconstrictive effect of sympathomimetic decongestants.
The parasympathetic innervation of the nasal turbinates is delivered through nerve fibers that reach the nasal turbinates through the posterior nasal nerve which crosses the sphenopalatine foramen and distributes to the mucosa following the branches of the sphenopalatine vessels ( 68 ). The result is vasodilation of erectile tissue in the nasal turbinates obstructing CSF lymphatic drainage.
10.3 Increased nasal blood pooling in patients with metabolic syndrome
The authors recently published an article that described subjects with metabolic syndrome, including hypertension, increased BMI, diabetes, and sleep apnea, exhibiting significantly increased nasal blood volume (2–3-fold greater), also referred to as blood pooling, as compared with subjects without metabolic syndrome as determined by whole-body nuclear imaging ( 25 ). This unique phenomenon of nasal pooling has been observed by the author using scintigraphic whole-body imaging in patients with metabolic syndrome, regardless of their body habitus ( Figure 3 ).

Figure 3 . Nuclear medicine scans of patients with metabolic syndrome.
Figure 3 shows increased nasal blood pooling in both patients with metabolic syndrome, including essential hypertension. Patient A, a 59-year-old female with a BMI of 32.5, has less severe metabolic syndrome. She has hypertension and hyperlipidemia with high triglycerides and is being treated with 1 anti-hypertensive and 1 anti-hyperlipidemic medication. Patient B has more severe metabolic syndrome with a BMI of 43.3. The patient is a 55-year-old female with hypertension, diabetes, sleep apnea, and hyperlipidemia, and is being treated with 3 anti-hypertensive, 1 anti-hyperlipidemic, and 1 anti-hyperglycemic medication.
As in patients with hypertension and without complete metabolic syndrome, we hypothesize that those patients with metabolic syndrome have an increase in blood volume in their nasal region significantly decreasing the normal lymphatic transport or drainage through the nasal turbinates resulting in increased intracranial pressure. The increased intracranial pressure causes the increased systemic blood pressure as part of Cushing’s mechanism, i.e., the selfish brain’s attempt to maintain cerebral blood flow. Patient A, the patient on the left in Figure 3 , has a less severe form of metabolic syndrome (without diabetes). She, however, has three of the five criteria of metabolic syndrome, including triglycerides over 150, a high waist circumference, and high blood pressure. Nonetheless, she has a high nose/heart ratio.
11 Controversy over which comes first, essential hypertension or increased brain blood flow resistance
There continues to be controversy in this area with most researchers believing that it is the development of essential hypertension from a mechanism not related to the brain that occurs first and leads to increased resistance to brain blood flow. Far fewer researchers believe that the brain is involved in the initiation of hypertension as proposed by Jennings et al. ( 69 , 70 ), or that an initial increase in resistance of brain blood flow leads to the development of essential hypertension as proposed by Dickinson and Thomason ( 71 ), Paton et al. ( 30 ), and Hart ( 14 ).
To understand the relationship between decreased nasal drainage, increased intracranial pressure, and hypertension, one must first be familiar with the normal nasal cycle.
12 CSF lymphatics and the normal nasal cycle
The nasal cycle is the alternating of airflow between nostrils that shifts between the left and right sides over time ( 72 ). The physical mechanism causing the nasal cycle is due to an asymmetry in blood flow leading to the engorgement of erectile tissue in the inferior turbinate and the anterior part of the nasal septum in one nostril more than the other. This normal asymmetrical enlargement of a nasal turbinate on one side blocks the passage of air. The autonomic nervous system mechanism is important in controlling the nasal cycle with sympathetic dominance associated with vasoconstriction and decongestion in one nostril while simultaneous parasympathetic vasodilation and congestion occur in the other nostril ( 72 ).
The purpose of the nasal cycle has been debated. Some studies suggest that the nasal cycle is a method of air conditioning and for removing entrapped contaminants ( 73 ). Eccles has proposed that the nasal cycle is a mechanism of respiratory defense against infection with respiratory viruses ( 74 ). Others have proposed that the nasal cycle could be a way to squeeze interstitial fluid out of the nasal turbinates during the constriction phase of the nasal cycle.
Although it has not been proposed that the nasal cycle serves as a pump to move lymphatic fluid from the CSF into the head and neck lymphatics, the authors believe that this could be one of the most important functions of the nasal cycle.
A malfunction of this normal cycle, with near-permanent vasodilation of the nasal erectile tissue, would result in a blockage of lymphatic outflow from the brain. In this regard, it is interesting that the nasal cycle was found to be diminished with age ( 75 , 76 ). In one study, 50% of patients over the age of 70 showed no evidence of a nasal cycle ( 76 ). Following thorough research, the authors were unable to find any current studies examining the effect of hypertension and metabolic syndrome on the nasal cycle. In our nuclear imaging studies of the blood pool, we did not visualize any asymmetry in the distribution of blood in the region of the nasal turbinates. Patients with hypertension in our whole-body blood pool imaging study, who also had a CT scan of the head, demonstrated symmetrically dilated right and left nasal turbinates without evidence of a nasal cycle (unpublished observation).
It is important to understand how CSF lymphatics are cleared from the brain.
13 Clearance of CSF from the brain
There has been considerable controversy regarding the most important pathway of clearance of CSF from the brain. For many years, the most accepted theory was that CSF was absorbed by the arachnoid granulations directly into the venous system. This theory has been significantly challenged over the last 40 years as many investigators have shown the importance of lymphatic clearance of CSF, primarily through the cribriform plate into the nasal region. In addition, a recent study using magnetic resonance imaging (MRI) provided evidence that a portion of the CSF is cleared by the parenchymal venous system ( 77 ) with only minimal contribution of the arachnoid granulations in CSF clearance. Further studies are required to provide a better understanding of the contribution of CSF lymphatics, the parenchymal venous system, and arachnoid granulation to overall CSF clearance, however, there has been increasing evidence for the importance of nasal lymphatics in CSF clearance ( 78 – 84 ).
14 Evidence for significant clearance of CSF through nasal lymphatics
A major proponent of this idea was Johnston et al. ( 78 , 82 , 85 ) whose work contradicted the most accepted theory that the majority of CSF is cleared by the arachnoid granulations. As pointed out by Johnston and Papaiconomou ( 79 ), there has been very limited evidence to support the idea that the arachnoid granulations are the primary site of CSF clearance from the brain; however, there has been significant research supporting clearance of CSF through the cribriform plate into the nasal turbinate region. In one study, Johnston’s group found that 30 min after injection of radiolabeled human serum albumin into the CSF, the tissue that contained the highest activity was the middle nasal turbinate which had approximately 6 times more activity than the blood ( 82 ). In another study, Johnston et al. ( 86 ) reported that approximately one-half of a protein tracer was transported from the CSF to the blood via extracranial lymphatic vessels. In another study by this group, when CSF transport was blocked through the cribriform plate, resting intracranial pressure doubled from 9.2 cmH 2 O to 18.0 cm H 2 O ( 87 ). A recent review of the importance of nasal lymphatics in CSF clearance has been published and is titled, “The brain-nose interface: a potential cerebrospinal fluid clearance site in humans” ( 80 ).
Since an original report by Schwalbe ( 88 ) in 1869, a large body of work in many different species has indicated a role for lymphatic vessels draining CSF in both cranial and spinal regions. However, only recently published anatomical and quantitative studies have shown that connections between the CSF and the extracranial lymphatic system represent a significant route for CSF drainage ( 83 , 84 , 89 , 90 ).
A PET imaging study by de Leon et al. ( 89 ) showed tracer activity in the nasal turbinates suggesting CSF movement through the cribriform plate and into the nasal turbinate lymphatics. In a recent study by Zhou et al. ( 83 ), 92 patients clearly showed activity in the inferior nasal turbinates following intrathecal infusion of an MRI contrast agent. Another recent 2023 study in rats using high-resolution imaging was strongly supportive of lymphatic movement along olfactory nerves. The study concluded that the olfactory nerve pathway into nasal turbinate lymphatics is the major route of CSF clearance from the brain ( 90 ).
In another animal model study, infusion of Ringer’s lactate with blue dye into the cisterna magna to increase the intracranial pressure caused a 3-fold increase in cervical lymph node flow and an increase in blue-colored nasal discharge that appeared 48 min after the beginning of the infusion ( 91 ). The nasal discharge increased from negligible, before the cisternal infusion, to 11.4 mL/h following the infusion. These studies support the clearance of CSF in cervical lymphatics and nasal fluid.
Ma et al. ( 92 ) found that lymphatic vessels were the major outflow pathway of CSF for both large and small molecular tracers in mice. They also found a significant decline in CSF lymphatic outflow in aged compared to young mice suggesting that the lymphatic system may represent a target for age-associated neurological conditions. In another recent study by Yoon et al. ( 84 ), a nasopharyngeal lymphatic plexus was found to be a hub for CSF drainage to the deep cervical lymph nodes. This plexus was suggested as a possible target for the treatment of age-related neurological conditions which are known to be associated with decreased CSF transport to deep cervical lymph nodes.
Meningeal lymphatic vessels located along the dural sinuses have been shown to drain into the cervical lymph nodes ( 93 ), and are coupled with, and receive drainage from, the recently described glymphatic system within the brain ( 94 ) that was first described by Iliff et al. ( 95 ) in 2012 and which will be discussed in the next section.
15 The glymphatic/lymphatic system
15.1 the glymphatic system.
The glymphatic system consists of specialized low-resistance spaces known as Virchow–Robin paravascular spaces that permit CSF inflow deep into the neural parenchyma. A detailed review of this glymphatic system has recently been published by the author (WP) and colleagues ( 96 ). The glymphatic system runs in the same direction as blood flow which is propelled by pulsations from the arterial vascular wall. This system can deliver protective molecules, such as melatonin, deep into the brain along the periarterial spaces. It also transports protein waste products, such as amyloid and tau degradation products, from the brain via the paravenous spaces ( 97 ). The fluid in the paravenous space eventually moves into the subarachnoid space on the surface of the brain where the fluid and any waste material are absorbed into meningeal lymphatic vessels as reported by Aspelund et al. ( 98 ) and Louveau et al. ( 99 ) in 2015. This network of meningeal lymphatics serves the same purpose as classical lymphatic drainage and is essential for maintaining neurophysiological homeostasis. The fluid in the meningeal lymphatics is then transported out of the brain and moves into cervical lymphatics. Although the precise anatomic pathway taken by this CSF/lymphatic fluid out of the cranial cavity remains to be clearly defined, the greatest evidence supports its movement along the cranial and spinal nerves, with the olfactory nerve thought to be the most predominant ( 78 , 82 ). Drainage from these meningeal and cervical lymphatics is relatively fast as tracers injected into the brain or CSF accumulate in the cervical lymph nodes within minutes after injection into the brain or CSF ( 100 ). The discovery of this glymphatic/lymphatic clearance system has clearly shown that CSF and interstitial fluid are directionally transported within the CNS.
Interestingly, it has been shown that this glymphatic/lymphatic clearance system is impaired with age at which time hypertension also develops ( 101 ). Because the glymphatic/lymphatic system plays a key role in the clearance of amyloid-beta and tau proteins, this system has been suggested to represent a new target to combat neurodegenerative disease ( 102 ). There is a recent MRI tracer imaging study supporting this theory which showed that impaired peri-olfactory cerebrospinal fluid clearance through the inferior turbinate was associated with aging, cognitive decline, and decreased sleep quality ( 83 ).
15.2 Importance of lymphatic nasal drainage for brain fluid homeostasis
Abnormally increased parasympathetic-induced nasal turbinate vasodilation and resultant blood pooling that interferes with the normal nasal cycle would be expected to obstruct lymphatic flow from the brain. In a rat model, nasal turbinate lymphatics were shown to be important for the clearance of CNS fluid when intracranial pressure was artificially increased ( 85 ). An increase in intracranial pressure by infusion of plasma into the lateral ventricle resulted in elevated pressure in the deep cervical lymph nodes which receive lymphatic drainage from the nasal turbinates. Very recently reported research in a rat model has also shown that CSF moves through the cribriform plate along the olfactory nerve to join lymphatics in the nasal mucosa which then are carried to a nasopharyngeal lymphatic plexus. CSF then drains to cervical lymph nodes through medial deep cervical lymphatics. These medial deep cervical lymphatics carry a significantly greater volume of CSF as compared to the lateral deep cervical lymphatics ( 84 ).
16 Increased intracranial pressure consistent with arterial wall thickening
In previous studies, narrowing and thickening of the cervical arteries feeding the brain were cited as evidence of the selfish brain hypothesis of hypertension. The theory is that the vessel narrowing, caused either congenitally or due to atheroma formation, causes an elevation of blood pressure as the brain ensures that it has sufficient blood flow through these narrowed arteries acting via Cushing’s mechanism ( 14 , 30 , 31 ). Vertebral artery thickening has been shown to occur in spontaneously hypertensive rats (SHR) before the development of systemic hypertension ( 30 ) supporting the selfish brain theory.
We believe another possible explanation for the thickened vessel walls is that increased intracranial pressure causes a back pressure in the arteries feeding the brain which leads to thickening of the cervical arteries. We hypothesize that lymphatic obstruction of CSF outflow through the nasal turbinates causes increased intracranial pressure and it is this increased intracranial pressure that leads to vessel wall thickening and increased systemic blood pressure as part of the selfish brain hypothesis, i.e., Cushing’s mechanism.
Evidence compatible with increased vascular thickening due to lymphatic obstruction has recently been published in the case of lymphedema of the arms in which brachial arteries feeding the lymphedema arm have significantly greater thickening of the arterial walls compared to the non-lymphedematous arm ( 103 ). The significantly increased wall thickness was principally due to increased intima-media thickening resulting in 0.38 mm in the lymphedema arm versus 0.31 mm in the normal arm ( p = 0.0008).
17 Glymphatic/lymphatic obstruction of CSF leads to increased intracranial pressure and hypertension
The obstruction of CSF lymphatic clearance from the brain at the level of the nasal turbinate due to abnormal turbinate vasodilation and blood pooling would result in decreased clearance of lymphatic fluid from the brain. The decrease in lymphatic fluid drainage would also decrease glymphatic function and the clearance of CSF waste proteins from the brain. Evidence of fluid obstruction in the glymphatic space is provided by MRI imaging in which the perivascular space has been noted to be enlarged in patients with hypertension ( 104 ). Evidence of the coupling of the glymphatic system to meningeal lymphatics was first described by Louveau et al. ( 99 ) using fluorescent markers. Evidence has also been found for the coupling of these two systems in humans using MRI imaging ( 94 ). Prior studies by Johnston ( 78 ) and recent PET and MRI imaging studies have shown that significant CSF clearance passes through the nasal turbinates ( 83 , 89 ). The decreased clearance of fluid from brain lymphatics and the glymphatic system due to nasal turbinate lymphatic obstruction would lead to increased intracranial pressure and a subsequent increase in the systemic blood pressure required to maintain normal blood flow to the brain via Cushing’s mechanism ( Figure 4 ).

Figure 4 . Illustration of consequences of nasal turbinate obstruction leading to increased intracranial pressure and resultant essential hypertension.
Mildly increased intracranial pressure due to lymphatic obstruction would also explain the significant correlation our group observed with increased nasal blood pooling and headaches (unpublished observations to be submitted soon). Similarly, the vasodilation of nasal erectile tissue caused by sildenafil, a drug commonly used to treat erectile dysfunction, causes symptomatic nasal obstruction and headaches ( 105 ). Sildenafil is one of the most common causes of drug-induced headaches ( 106 ).
18 Sleep disturbances
18.1 the importance of sleep for adequate csf lymphatic drainage.
Numerous associations have been documented between sleep disturbances and the failure to clear waste products from the brain ( 107 ). Sleep disturbances are associated with increased CSF metabolite concentrations (e.g., amyloid-beta, orexin, tau proteins), and increased CSF volumes or pressure ( 108 ). Recent studies have suggested that glymphatic dysfunction is a common underlying etiology of sleep disorders and headache pain ( 109 ). The glymphatic system is particularly active during sleep whereby potentially toxic neural waste substances that accumulate during wakefulness are cleared via the glymphatic system ( 108 , 110 ). It is thought that the brain cell volume decreases during sleep, expanding the size of the paravascular space, and facilitating the influx of CSF into the peritubular space for material exchange and metabolic waste removal ( 111 ). Animal experiments using intravital 2-photon microscopy in mice showed that glymphatic clearance is decreased by 90% during wakefulness, while protein clearance in the intima of the brain doubles during sleep ( 97 , 112 ).
Short sleep duration has also been associated with essential hypertension in many epidemiologic studies ( 113 ), although there has been no clear pathophysiologic connection found between the two. It is the authors’ hypothesis that decreased CSF clearance due to short sleep and obstructed nasal lymphatics is related to the development of hypertension.
18.2 Sleep apnea and hypertension
Obstructive sleep apnea (OSA), another common disorder, is strongly associated with the development of hypertension ( 114 – 116 ) and recent evidence suggests that it may also be linked with cognitive decline and dementia. Sleep apnea may be the common pathway linking hypertension and the development of dementia ( 117 ).
The authors have found significantly increased nasal turbinate blood pooling in patients with OSA. In our review of 200 patients, sleep apnea patients had an average nose-to-heart max ratio of 0.99 versus 0.86 in patients without sleep apnea ( p = 0.0002) using the Wilcoxon rank-sum test ( 25 ). An example image of a patient with sleep apnea is shown in Figure 5 . Subjects with OSA have also been shown to have increased sympathetic activity and sleep apnea has been linked to resistant hypertension ( 118 ). Although not yet investigated, based on our findings of increased nasal turbinate vasodilation in patients with OSA, it is likely that these sleep apnea patients also have increased nasal parasympathetic activity. In this regard, a recently published Phase 2 study has shown that a drug that reduces the activity of the parasympathetic system significantly improves OSA ( 119 ). This drug, taken before bedtime, significantly reduced the apnea-hypopnea index in OSA patients.

Figure 5 . Patients without (A) and with (B) sleep apnea.
A prior study in sleep apnea patients used CT-acquired nasal turbinate measurements to find a significantly positive correlation between the size of the inferior nasal turbinates in obese patients with sleep apnea ( 120 ). This prior study did not include patients without sleep apnea so there were no direct comparisons of nasal turbinate size between sleep apnea patients and normal subjects; however, future studies using this CT methodology could be performed to investigate the correlation of nasal turbinate dimensions with hypertension and sleep apnea in the future.
Figure 5 is a nuclear scan of a 49-year-old female (Patient A) without sleep apnea, hypertension, hyperlipidemia, or diabetes, with a nose/heart max ratio of 0.67. Patient B is a 52-year-old female with sleep apnea but without hypertension or diabetes at the time of the study, with a nose/heart max ratio of 1.16. A CT scan at the time of the nuclear study showed dilated nasal turbinates. A 3-year follow-up scan of Patient B showed an increased nose/heart max ratio of 1.28. The patient had developed Stage 1 hypertension and pre-diabetes.
Increased intracranial pressure has been associated with sleep apnea ( 121 , 122 ), a known risk factor for hypertension ( 123 ). Treatment of sleep apnea has been suggested as a method to prevent hypertension ( 124 ). The increased intracranial pressure associated with sleep apnea and obesity has even been reported to cause thinning of the skull with an increased likelihood of producing a CSF leak ( 122 ). Our findings of nasal vasodilation in patients with hypertension and sleep apnea suggest the possibility that obstruction of the CSF lymphatic clearance from the brain through nasal turbinate lymphatics is responsible for the increased intracranial pressure and the resultant sleep apnea and hypertension. Obstruction of nasal turbinate lymphatic flow as described in this article could also be related to the development of idiopathic intracranial hypertension (IIH). The most common occurrence of IIH is in obese women of childbearing age who are also more likely to have essential hypertension and metabolic syndrome. Sleep apnea has also been associated with both essential hypertension and IIH ( 116 , 125 ).
19 Future confirmatory studies
The proposed mechanism of essential hypertension presented in this paper is a working hypothesis and confirmatory studies will be needed. There is a potential for prospective studies to complement the retrospective studies in patients with hypertension as discussed in this paper. Studies could be performed using nuclear blood pool imaging to assess nasal turbinates as in our retrospective studies. An advantage of the nuclear imaging technique is that dynamic imaging, viewing changes in the nasal blood pool over 8 h, could be performed by simply placing a standard gamma camera over the upper body of the patient. This would allow studies to be performed during sleep or during other medical or physical interventions that affect the nasal turbinates. The gamma camera could be placed several inches away from the patient, resulting in minimal disturbance. To perform prolonged studies, a blood pool imaging agent such as labeled red blood cells could be utilized to permit dynamic imaging for 8–12 h. Technetium-99 m labeled red blood cells are standard blood pool nuclear imaging agents most commonly used for locating the site of gastrointestinal bleeding, diagnosing hepatic hemangiomas, and determining left ventricular ejection fractions ( 126 ). Other imaging studies to assess nasal turbinate vasodilation in hypertension could be performed with MRI or CT, such as those previously reported by Rodrigues et al. ( 120 ), stating that obese patients had inferior turbinate hypertrophy.
Other studies could be performed with MRI contrast agents, investigating the lymphatic drainage of cerebrospinal fluid through nasal turbinates and its association with hypertension. Studies could also be performed to further assess the absence or presence of the nasal cycle in patients with hypertension as compared to controls.
20 Potential novel therapeutic approaches targeting the nasal turbinates
Based on the evidence in this paper, the nasal turbinates are potential targets for the treatment of hypertension. One possible treatment would be to block the increased parasympathetic activity of the nasal turbinates by blocking the sphenopalatine ganglion that carries parasympathetic activity to the nasal turbinates. The sphenopalatine ganglion is the largest extracranial parasympathetic ganglion of the head ( 127 ). Sphenopalatine ganglion blockage has been used to treat migraine headaches ( 127 ) and a recent study has shown that blocking the sphenopalatine ganglion can modestly lower blood pressure ( 128 ). However, completely blocking parasympathetic activity to the nose may not be the best approach for treating hypertension as it would adversely affect the nasal cycle which is dependent on alternating sympathetic and parasympathetic activity to the nasal turbinates ( 72 ) which may be important for the clearance of CSF fluid from the brain.
Future therapeutic approaches could be aimed at increasing the volume of CSF flowing through the nasal lymphatics. These therapies’ goal would be to restore the nasal cycle or to use other physical approaches to increase the movement of CSF through and out of the brain. Decreasing intracranial pressure through therapy targeted at the nasal turbinates could lead to significantly improved blood pressure control and a more effective treatment for sleep apnea.
21 Conclusion
Finding more effective treatments for essential hypertension offers the possibility of better blood pressure control resulting in a decrease in the incidence of myocardial infarction, stroke, renal failure, dementia, and overall mortality currently associated with hypertension. Considering that accumulation of amyloid and tau proteins in the brain are involved in the pathogenesis of neurodegenerative diseases, the potential of treating hypertension by methods that focus on nasal turbinate obstruction and/or increasing cerebrospinal fluid lymphatic flow may also offer a therapeutic benefit for neurodegenerative pathologies in addition to its potential to treat hypertension.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Author contributions
WP: Conceptualization, Methodology, Writing – original draft, Writing – review & editing. JS: Conceptualization, Methodology, Writing – original draft, Writing – review & editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
The authors thank Jonathan Sumner for his contribution to the medical illustration ( Figure 4 ).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. GBD 2019 Risk Factors Collaborators. Global burden of 87 risk factors in 204 countries and territories, 1990–2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet . (2020) 396:1223–49. doi: 10.1016/S0140-6736(20)30752-2
PubMed Abstract | Crossref Full Text | Google Scholar
2. NCD Risk Factor Collaboration (NCD-RisC). Worldwide trends in hypertension prevalence and progress in treatment and control from 1990 to 2019: a pooled analysis of 1201 population-representative studies with 104 million participants. Lancet . (2021) 398:957–80. doi: 10.1016/S0140-6736(21)01330-1
3. Mills, KT, Stefanescu, A, and He, J. The global epidemiology of hypertension. Nat Rev Nephrol . (2020) 16:223–37. doi: 10.1038/s41581-019-0244-2
4. Fryar, CD, Carroll, MD, and Ogden, CL. Prevalence of overweight, obesity, and extreme obesity among adults: United States, 1960–1962 through 2011–2012 . Hyattsville, Maryland, USA: National Center for Health Statistics (2014).
Google Scholar
5. Pan, XF, Wang, L, and Pan, A. Epidemiology and determinants of obesity in China. Lancet Diabetes Endocrinol . (2021) 9:373–92. doi: 10.1016/S2213-8587(21)00045-0
Crossref Full Text | Google Scholar
6. Bludorn, J, and Railey, K. Hypertension guidelines and interventions. Prim Care . (2024) 51:41–52. doi: 10.1016/j.pop.2023.07.002
7. Ouyang, H. Bariatric surgery at 16. N Y Times Mag . (2023):24–33.
8. Tu, WJ, Zhao, Z, Yin, P, Cao, L, Zeng, J, Chen, H, et al. Estimated burden of stroke in China in 2020. JAMA Netw Open . (2023) 6:e231455. doi: 10.1001/jamanetworkopen.2023.1455
9. Ferdinand, KC, Charbonnet, RM, Laurent, J, and Villavaso, CD. Eliminating hypertension disparities in U.S. non-Hispanic black adults: current and emerging interventions. Curr Opin Cardiol . (2023) 38:304–10. doi: 10.1097/HCO.0000000000001040
10. Onwuzo, C, Olukorode, JO, Omokore, OA, Odunaike, OS, Omiko, R, Osaghae, OW, et al. DASH diet: a review of its scientifically proven hypertension reduction and health benefits. Cureus . (2023) 15:e44692. doi: 10.7759/cureus.44692
11. Gupta, DK, Lewis, CE, Varady, KA, Su, YR, Madhur, MS, Lackland, DT, et al. Effect of dietary sodium on blood pressure: a crossover trial. JAMA . (2023) 330:2258–66. doi: 10.1001/jama.2023.23651
12. Grassi, G. Sympathetic modulation as a goal of antihypertensive treatment: from drugs to devices. J Hypertens . (2023) 41:1688–95. doi: 10.1097/HJH.0000000000003538
13. Carthy, ER. Autonomic dysfunction in essential hypertension: a systematic review. Ann Med Surg . (2014) 3:2–7. doi: 10.1016/j.amsu.2013.11.002
14. Hart, EC. Human hypertension, sympathetic activity and the selfish brain. Exp Physiol . (2016) 101:1451–62. doi: 10.1113/EP085775
15. Grassi, G, Dell’Oro, R, Quarti-Trevano, F, Vanoli, J, and Oparil, S. Sympathetic neural mechanisms in hypertension: recent insights. Curr Hypertens Rep . (2023) 25:263–70. doi: 10.1007/s11906-023-01254-4
16. Mancia, G, and Grassi, G. The autonomic nervous system and hypertension. Circ Res . (2014) 114:1804–14. doi: 10.1161/CIRCRESAHA.114.302524
17. Louca, P, Menni, C, and Padmanabhan, S. Genomic determinants of hypertension with a focus on metabolomics and the gut microbiome. Am J Hypertens . (2020) 33:473–81. doi: 10.1093/ajh/hpaa022
18. Calvillo, L, Gironacci, MM, Crotti, L, Meroni, PL, and Parati, G. Neuroimmune crosstalk in the pathophysiology of hypertension. Nat Rev Cardiol . (2019) 16:476–90. doi: 10.1038/s41569-019-0178-1
19. Arif, M, Sadayappan, S, Becker, RC, Martin, LJ, and Urbina, EM. Epigenetic modification: a regulatory mechanism in essential hypertension. Hypertens Res . (2019) 42:1099–113. doi: 10.1038/s41440-019-0248-0
20. Fernandes-Rosa, FL, Boulkroun, S, Fedlaoui, B, Hureaux, M, Travers-Allard, S, Drossart, T, et al. New advances in endocrine hypertension: from genes to biomarkers. Kidney Int . (2023) 103:485–500. doi: 10.1016/j.kint.2022.12.021
21. Rossi, GP, Ceolotto, G, Caroccia, B, and Lenzini, L. Genetic screening in arterial hypertension. Nat Rev Endocrinol . (2017) 13:289–98. doi: 10.1038/nrendo.2016.196
22. Micaglio, E, Locati, ET, Monasky, MM, Romani, F, Heilbron, F, and Pappone, C. Role of pharmacogenetics in adverse drug reactions: an update towards personalized medicine. Front Pharmacol . (2021) 12:651720. doi: 10.3389/fphar.2021.651720
23. Luft, FC. Did you know? Why is essential hypertension essential-or is it? Acta Physiol . (2020) 229:e13469. doi: 10.1111/apha.13469
24. Chan, RJ, Helmeczi, W, and Hiremath, SS. Revisiting resistant hypertension: a comprehensive review. Intern Med J . (2023) 53:1739–51. doi: 10.1111/imj.16189
25. Phillips, WT, Issa, NJ, Elhalwagi, SB, Draeger, HT, Schwartz, JG, and Gelfond, JA. Nasal and parotid blood pool activity is significantly correlated with metabolic syndrome components and sleep apnea. Metab Syndr Relat Disord . (2022) 20:395–404. doi: 10.1089/met.2022.0015
26. Phillips, WT, Salman, UA, McMahan, CA, and Schwartz, JG. Accelerated gastric emptying in hypertensive subjects. J Nucl Med . (1997) 38:207–11.
27. Phillips, WT, Schwartz, JG, and McMahan, CA. Rapid gastric emptying in patients with early non-insulin-dependent diabetes mellitus. N Engl J Med . (1991) 324:130–1. doi: 10.1056/nejm199101103240217
28. Schwartz, JG, Green, GM, Guan, D, McMahan, CA, and Phillips, WT. Rapid gastric emptying of a solid pancake meal in type II diabetic patients. Diabetes Care . (1996) 19:468–71. doi: 10.2337/diacare.19.5.468
29. Schwartz, JG, McMahan, CA, Green, GM, and Phillips, WT. Gastric emptying in Mexican Americans compared to non-Hispanic whites. Dig Dis Sci . (1995) 40:624–30. doi: 10.1007/bf02064382
30. Paton, JF, Dickinson, CJ, and Mitchell, G. Harvey Cushing and the regulation of blood pressure in giraffe, rat and man: introducing ‘Cushing’s mechanism’. Exp Physiol . (2009) 94:11–7. doi: 10.1113/expphysiol.2008.043455
31. Dickinson, CJ, and Thomson, AD. A post mortem study of the main cerebral arteries with special reference to their possible role in blood pressure regulation. Clin Sci . (1960) 19:513–38.
PubMed Abstract | Google Scholar
32. da Costa, MM, Sousa, ALL, Correia, MC, Inuzuka, S, Costa, TO, Vitorino, PVO, et al. Intracranial pressure waveform in patients with essential hypertension. Front Cardiovasc Med . (2023) 10:1288080. doi: 10.3389/fcvm.2023.1288080
33. Dickinson, CJ. Reappraisal of the Cushing reflex: the most powerful neural blood pressure stabilizing system. Clin Sci . (1990) 79:543–50. doi: 10.1042/cs0790543
34. Kety, SS, Hafkenschiel, JH, Jeffers, WA, Leopold, IH, and Shenkin, HA. The blood flow, vascular resistance, and oxygen consumption of the brain in essential hypertension. J Clin Invest . (1948) 27:511–4. doi: 10.1172/jci101998
35. Paton, JF, and Raizada, MK. Neurogenic hypertension. Exp Physiol . (2010) 95:569–71. doi: 10.1113/expphysiol.2009.047282
36. Fisher, JP, and Paton, JF. The sympathetic nervous system and blood pressure in humans: implications for hypertension. J Hum Hypertens . (2012) 26:463–75. doi: 10.1038/jhh.2011.66
37. Cushing, H. Concerning a definitive regulatory mechanism of the vasomotor centre which controls blood pressure during cerebral compression. Bull Johns Hopk Hosp . (1901):290–2.
38. Cushing, H. Some experimental and clinical observations concerning states of increased intracranial tension. Am J Med Sci . (1902) 124:375–400. doi: 10.1097/00000441-190209000-00001
39. Smith, ER, and Madsen, JR. Cerebral pathophysiology and critical care neurology: basic hemodynamic principles, cerebral perfusion, and intracranial pressure. Semin Pediatr Neurol . (2004) 11:89–104. doi: 10.1016/j.spen.2004.04.001
40. Nobili, F, Rodriguez, G, Marenco, S, De Carli, F, Gambaro, M, Castello, C, et al. Regional cerebral blood flow in chronic hypertension. A correlative study. Stroke . (1993) 24:1148–53. doi: 10.1161/01.str.24.8.1148
41. Faraci, FM, and Heistad, DD. Regulation of large cerebral arteries and cerebral microvascular pressure. Circ Res . (1990) 66:8–17. doi: 10.1161/01.res.66.1.8
42. Schmidt, EA, Despas, F, Pavy-Le Traon, A, Czosnyka, Z, Pickard, JD, Rahmouni, K, et al. Intracranial pressure is a determinant of sympathetic activity. Front Physiol . (2018) 9:11. doi: 10.3389/fphys.2018.00011
43. Warnert, EA, Rodrigues, JC, Burchell, AE, Neumann, S, Ratcliffe, LE, Manghat, NE, et al. Is high blood pressure self-protection for the brain? Circ Res . (2016) 119:e140–51. doi: 10.1161/CIRCRESAHA.116.309493
44. Manghat, NE, Robinson, E, Mitrousi, K, Rodrigues, JCL, Hinton, T, Paton, JFR, et al. Cerebrovascular variants and the role of the selfish brain in young-onset hypertension. Hypertension . (2022) 79:1265–74. doi: 10.1161/HYPERTENSIONAHA.121.18612
45. Bhatia, V, and Tandon, RK. Stress and the gastrointestinal tract. J Gastroenterol Hepatol . (2005) 20:332–9. doi: 10.1111/j.1440-1746.2004.03508.x
46. Baraniuk, JN, and Merck, SJ. New concepts of neural regulation in human nasal mucosa. Acta Clin Croat . (2009) 48:65–73. doi: 10.1111/j.1749-6632.2009.04481.x
47. Armstrong, R, Wheen, P, Brandon, L, Maree, A, and Kenny, RA. Heart rate: control mechanisms, pathophysiology and assessment of the neurocardiac system in health and disease. QJM . (2022) 115:806–12. doi: 10.1093/qjmed/hcab016
48. Esler, M. The sympathetic nervous system in hypertension: back to the future? Curr Hypertens Rep . (2015) 17:11. doi: 10.1007/s11906-014-0519-8
49. Grassi, G, Biffi, A, Dell’Oro, R, Quarti Trevano, F, Seravalle, G, Corrao, G, et al. Sympathetic neural abnormalities in type 1 and type 2 diabetes: a systematic review and meta-analysis. J Hypertens . (2020) 38:1436–42. doi: 10.1097/HJH.0000000000002431
50. Smith, PA, Graham, LN, Mackintosh, AF, Stoker, JB, and Mary, DA. Relationship between central sympathetic activity and stages of human hypertension. Am J Hypertens . (2004) 17:217–22. doi: 10.1016/j.amjhyper.2003.10.010
51. Wallin, BG, Delius, W, and Hagbarth, KE. Comparison of sympathetic nerve activity in normotensive and hypertensive subjects. Circ Res . (1973) 33:9–21. doi: 10.1161/01.res.33.1.9
52. Seravalle, G, and Grassi, G. Sympathetic nervous system and hypertension: new evidences. Auton Neurosci . (2022) 238:102954. doi: 10.1016/j.autneu.2022.102954
53. Morise, T, Horita, M, Kitagawa, I, Shinzato, R, Hoshiba, Y, Masuya, H, et al. The potent role of increased sympathetic tone in pathogenesis of essential hypertension with neurovascular compression. J Hum Hypertens . (2000) 14:807–11. doi: 10.1038/sj.jhh.1001114
54. Böhm, R, van Baak, M, van Hooff, M, Moy, J, and Rahn, KH. Salivary flow in borderline hypertension. Klin Wochenschr . (1985) 63:154–6.
55. Salman, UA, Schwartz, JG, McMahan, CA, Michalek, JE, and Phillips, WT. Rapid gastric emptying in spontaneously hypertensive rats. J Hypertens . (2023) 42:572–8. doi: 10.1097/HJH.0000000000003640
56. De Fano, M, Porcellati, F, Fanelli, CG, Corio, S, Mazzieri, A, Lucidi, P, et al. The role of gastric emptying in glucose homeostasis and defense against hypoglycemia: innocent bystander or partner in crime? Diabetes Res Clin Pract . (2023) 203:110828. doi: 10.1016/j.diabres.2023.110828
57. Murthy, TA, Grivell, J, Hatzinikolas, S, Chapple, LS, Chapman, MJ, Stevens, JE, et al. Acceleration of gastric emptying by insulin-induced hypoglycemia is dependent on the degree of hypoglycemia. J Clin Endocrinol Metab . (2021) 106:364–71. doi: 10.1210/clinem/dgaa854
58. Schvarcz, E, Palmér, M, Aman, J, and Berne, C. Hypoglycemia increases the gastric emptying rate in healthy subjects. Diabetes Care . (1995) 18:674–6. doi: 10.2337/diacare.18.5.674
59. Lopez-Gambero, AJ, Martinez, F, Salazar, K, Cifuentes, M, and Nualart, F. Brain glucose-sensing mechanism and energy homeostasis. Mol Neurobiol . (2019) 56:769–96. doi: 10.1007/s12035-018-1099-4
60. Shi, M, Jones, AR, Niedringhaus, MS, Pearson, RJ, Biehl, AM, Ferreira, M Jr, et al. Glucose acts in the CNS to regulate gastric motility during hypoglycemia. Am J Physiol Regul Integr Comp Physiol . (2003) 285:R1192–202. doi: 10.1152/ajpregu.00179.2003
61. Phillips, WT, Schwartz, JG, and McMahan, CA. Reduced postprandial blood glucose levels in recently diagnosed non-insulin-dependent diabetics secondary to pharmacologically induced delayed gastric emptying. Dig Dis Sci . (1993) 38:51–8. doi: 10.1007/BF01296773
62. Lincoff, AM, Brown-Frandsen, K, Colhoun, HM, Deanfield, J, Emerson, SS, Esbjerg, S, et al. Semaglutide and cardiovascular outcomes in obesity without diabetes. N Engl J Med . (2023) 389:2221–32. doi: 10.1056/NEJMoa2307563
63. Lovshin, JA, and Zinman, B. Blood pressure-lowering effects of incretin-based diabetes therapies. Can J Diabetes . (2014) 38:364–71. doi: 10.1016/j.jcjd.2014.05.001
64. Wang, B, Zhong, J, Lin, H, Zhao, Z, Yan, Z, He, H, et al. Blood pressure-lowering effects of GLP-1 receptor agonists exenatide and liraglutide: a meta-analysis of clinical trials. Diabetes Obes Metab . (2013) 15:737–49. doi: 10.1111/dom.12085
65. Li, QX, Gao, H, Guo, YX, Wang, BY, Hua, RX, Gao, L, et al. GLP-1 and underlying beneficial actions in Alzheimer’s disease, hypertension, and NASH. Front Endocrinol . (2021) 12:721198. doi: 10.3389/fendo.2021.721198
66. Cole, P, Haight, JS, Cooper, PW, and Kassel, EE. A computed tomographic study of nasal mucosa: effects of vasoactive substances. J Otolaryngol . (1983) 12:58–60.
67. Ng, BA, Ramsey, RG, and Corey, JP. The distribution of nasal erectile mucosa as visualized by magnetic resonance imaging. Ear Nose Throat J . (1999) 78:159–6. doi: 10.1177/014556139907800309
68. Cassano, M, Russo, L, Del Giudice, AM, and Gelardi, M. Cytologic alterations in nasal mucosa after sphenopalatine artery ligation in patients with vasomotor rhinitis. Am J Rhinol Allergy . (2012) 26:49–54. doi: 10.2500/ajra.2012.26.3683
69. Jennings, JR, Muldoon, MF, and Sved, AF. Is the brain an early or late component of essential hypertension? Am J Hypertens . (2020) 33:482–90. doi: 10.1093/ajh/hpaa038
70. Jennings, JR, and Zanstra, Y. Is the brain the essential in hypertension? NeuroImage . (2009) 47:914–21. doi: 10.1016/j.neuroimage.2009.04.072
71. Dickinson, CJ, and Thomason, AD. Vertebral and internal carotid arteries in relation to hypertension and cerebrovascular disease. Lancet . (1959) 2:46–8. doi: 10.1016/s0140-6736(59)90494-5
72. Kahana-Zweig, R, Geva-Sagiv, M, Weissbrod, A, Secundo, L, Soroker, N, and Sobel, N. Measuring and characterizing the human nasal cycle. PLoS One . (2016) 11:e0162918. doi: 10.1371/journal.pone.0162918
73. Soane, RJ, Carney, AS, Jones, NS, Frier, M, Perkins, AC, Davis, SS, et al. The effect of the nasal cycle on mucociliary clearance. Clin Otolaryngol Allied Sci . (2001) 26:9–15. doi: 10.1046/j.1365-2273.2001.00423.x
74. Eccles, R. The role of nasal congestion as a defence against respiratory viruses. Clin Otolaryngol . (2021) 46:4–8. doi: 10.1111/coa.13658
75. Mirza, N, Kroger, H, and Doty, RL. Influence of age on the ‘nasal cycle’. Laryngoscope . (1997) 107:62–6. doi: 10.1097/00005537-199701000-00014
76. Williams, MR, and Eccles, R. The nasal cycle and age. Acta Otolaryngol . (2015) 135:831–4. doi: 10.3109/00016489.2015.1028592
77. Hu, J, Shen, Y, Fahmy, LM, Krishnamurthy, S, Li, J, Zhang, L, et al. The role of the parenchymal vascular system in cerebrospinal fluid tracer clearance. Eur Radiol . (2023) 33:656–65. doi: 10.1007/s00330-022-09022-9
78. Johnston, M. The importance of lymphatics in cerebrospinal fluid transport. Lymphat Res Biol . (2003) 1:41–5. doi: 10.1089/15396850360495682
79. Johnston, M, and Papaiconomou, C. Cerebrospinal fluid transport: a lymphatic perspective. News Physiol Sci . (2002) 17:227–30. doi: 10.1152/nips.01400.2002
80. Mehta, NH, Sherbansky, J, Kamer, AR, Carare, RO, Butler, T, Rusinek, H, et al. The brain-nose interface: a potential cerebrospinal fluid clearance site in humans. Front Physiol . (2021) 12:769948. doi: 10.3389/fphys.2021.769948
81. Chae, J, Choi, M, Choi, J, and Yoo, SJ. The nasal lymphatic route of CSF outflow: implications for neurodegenerative disease diagnosis and monitoring. Anim Cells Syst . (2024) 28:45–54. doi: 10.1080/19768354.2024.2307559
82. Nagra, G, Koh, L, Zakharov, A, Armstrong, D, and Johnston, M. Quantification of cerebrospinal fluid transport across the cribriform plate into lymphatics in rats. Am J Physiol Regul Integr Comp Physiol . (2006) 291:R1383–9. doi: 10.1152/ajpregu.00235.2006
83. Zhou, Y, Ran, W, Luo, Z, Wang, J, Fang, M, Wei, K, et al. Impaired peri-olfactory cerebrospinal fluid clearance is associated with ageing, cognitive decline and dyssomnia. EBioMedicine . (2022) 86:104381. doi: 10.1016/j.ebiom.2022.104381
84. Yoon, JH, Jin, H, Kim, HJ, Hong, SP, Yang, MJ, Ahn, JH, et al. Nasopharyngeal lymphatic plexus is a hub for cerebrospinal fluid drainage. Nature . (2024) 625:768–77. doi: 10.1038/s41586-023-06899-4
85. Koh, L, Nagra, G, and Johnston, M. Properties of the lymphatic cerebrospinal fluid transport system in the rat: impact of elevated intracranial pressure. J Vasc Res . (2007) 44:423–32. doi: 10.1159/000104255
86. Boulton, M, Flessner, M, Armstrong, D, Hay, J, and Johnston, M. Lymphatic drainage of the CNS: effects of lymphatic diversion/ligation on CSF protein transport to plasma. Am J Phys . (1997) 272:R1613–9. doi: 10.1152/ajpregu.1997.272.5.R1613
87. Mollanji, R, Bozanovic-Sosic, R, Zakharov, A, Makarian, L, and Johnston, MG. Blocking cerebrospinal fluid absorption through the cribriform plate increases resting intracranial pressure. Am J Physiol Regul Integr Comp Physiol . (2002) 282:R1593–9. doi: 10.1152/ajpregu.00695.2001
88. Schwalbe, G. Die Arachnoidalraum ein Lymphraum und sein Zusammenhang mit den Perichorioidalraum. [the arachnoidal space as a lymphatic space with connection to the perichoroidal compartment]. Zbl Med Wiss . (1869) 7:4650467.
89. de Leon, MJ, Li, Y, Okamura, N, Tsui, WH, Saint-Louis, LA, Glodzik, L, et al. Cerebrospinal fluid clearance in Alzheimer disease measured with dynamic PET. J Nucl Med . (2017) 58:1471–6. doi: 10.2967/jnumed.116.187211
90. Spera, I, Cousin, N, Ries, M, Kedracka, A, Castillo, A, Aleandri, S, et al. Open pathways for cerebrospinal fluid outflow at the cribriform plate along the olfactory nerves. EBioMedicine . (2023) 91:104558. doi: 10.1016/j.ebiom.2023.104558
91. Leeds, SE, Kong, AK, and Wise, BL. Alternative pathways for drainage of cerebrospinal fluid in the canine brain. Lymphology . (1989) 22:144–6.
92. Ma, Q, Ineichen, BV, Detmar, M, and Proulx, ST. Outflow of cerebrospinal fluid is predominantly through lymphatic vessels and is reduced in aged mice. Nat Commun . (2017) 8:1434. doi: 10.1038/s41467-017-01484-6
93. Da Mesquita, S, Louveau, A, Vaccari, A, Smirnov, I, Cornelison, RC, Kingsmore, KM, et al. Functional aspects of meningeal lymphatics in ageing and Alzheimer’s disease. Nature . (2018) 560:185–91. doi: 10.1038/s41586-018-0368-8
94. Ringstad, G, and Eide, PK. Glymphatic-lymphatic coupling: assessment of the evidence from magnetic resonance imaging of humans. Cell Mol Life Sci . (2024) 81:131. doi: 10.1007/s00018-024-05141-2
95. Iliff, JJ, Wang, M, Liao, Y, Plogg, BA, Peng, W, Gundersen, GA, et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci Transl Med . (2012) 4:147ra111. doi: 10.1126/scitranslmed.3003748
96. Reiter, RJ, Sharma, R, Rosales-Corral, S, de Mange, J, Phillips, WT, Tan, DX, et al. Melatonin in ventricular and subarachnoid cerebrospinal fluid: its function in the neural glymphatic network and biological significance for neurocognitive health. Biochem Biophys Res Commun . (2022) 605:70–81. doi: 10.1016/j.bbrc.2022.03.025
97. Nedergaard, M, and Goldman, SA. Glymphatic failure as a final common pathway to dementia. Science . (2020) 370:50–6. doi: 10.1126/science.abb8739
98. Aspelund, A, Antila, S, Proulx, ST, Karlsen, TV, Karaman, S, Detmar, M, et al. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J Exp Med . (2015) 212:991–9. doi: 10.1084/jem.20142290
99. Louveau, A, Smirnov, I, Keyes, TJ, Eccles, JD, Rouhani, SJ, Peske, JD, et al. Structural and functional features of central nervous system lymphatic vessels. Nature . (2015) 523:337–41. doi: 10.1038/nature14432
100. Plog, BA, Dashnaw, ML, Hitomi, E, Peng, W, Liao, Y, Lou, N, et al. Biomarkers of traumatic injury are transported from brain to blood via the glymphatic system. J Neurosci . (2015) 35:518–26. doi: 10.1523/JNEUROSCI.3742-14.2015
101. Kress, BT, Iliff, JJ, Xia, M, Wang, M, Wei, HS, Zeppenfeld, D, et al. Impairment of paravascular clearance pathways in the aging brain. Ann Neurol . (2014) 76:845–61. doi: 10.1002/ana.24271
102. Mestre, H, Mori, Y, and Nedergaard, M. The brain’s glymphatic system: current controversies. Trends Neurosci . (2020) 43:458–66. doi: 10.1016/j.tins.2020.04.003
103. Klusch, V, Boyle, EC, Rustum, S, Franz, M, Park-Simon, TW, Haverich, A, et al. Chronic unilateral arm lymphedema correlates with increased intima-media thickness in the brachial artery. Vasa . (2022) 51:19–23. doi: 10.1024/0301-1526/a000982
104. Shulyatnikova, T, and Hayden, MR. Why are perivascular spaces important? Medicina . (2023) 59:917. doi: 10.3390/medicina59050917
105. Kiroglu, AF, Bayrakli, H, Yuca, K, Cankaya, H, and Kiris, M. Nasal obstruction as a common side-effect of sildenafil citrate. Tohoku J Exp Med . (2006) 208:251–4. doi: 10.1620/tjem.208.251
106. Young, WB. Drug-induced headache. Neurol Clin . (2004) 22:173–84. doi: 10.1016/s0733-8619(03)00090-2
107. Komaroff, AL. Does sleep flush wastes from the brain? JAMA . (2021) 325:2153–5. doi: 10.1001/jama.2021.5631
108. Chong, PLH, Garic, D, Shen, MD, Lundgaard, I, and Schwichtenberg, AJ. Sleep, cerebrospinal fluid, and the glymphatic system: a systematic review. Sleep Med Rev . (2022) 61:101572. doi: 10.1016/j.smrv.2021.101572
109. Yi, T, Gao, P, Zhu, T, Yin, H, and Jin, S. Glymphatic system dysfunction: a novel mediator of sleep disorders and headaches. Front Neurol . (2022) 13:885020. doi: 10.3389/fneur.2022.885020
110. Rasmussen, MK, Mestre, H, and Nedergaard, M. Fluid transport in the brain. Physiol Rev . (2022) 102:1025–151. doi: 10.1152/physrev.00031.2020
111. Xie, L, Kang, H, Xu, Q, Chen, MJ, Liao, Y, Thiyagarajan, M, et al. Sleep drives metabolite clearance from the adult brain. Science . (2013) 342:373–7. doi: 10.1126/science.1241224
112. Miyakoshi, LM, Staeger, FF, Li, Q, Pan, C, Xie, L, Kang, H, et al. The state of brain activity modulates cerebrospinal fluid transport. Prog Neurobiol . (2023) 229:102512:102512. doi: 10.1016/j.pneurobio.2023.102512
113. Killick, R, Stranks, L, and Hoyos, CM. Sleep deficiency and cardiometabolic disease. Sleep Med Clin . (2023) 18:331–47. doi: 10.1016/j.jsmc.2023.05.012
114. Battaglia, E, Banfi, P, Compalati, E, Nicolini, A, Diaz, DETT, Gonzales, M, et al. The pathogenesis of OSA-related hypertension: what are the determining factors? Minerva Med . (2023) 115:68–82. doi: 10.23736/s0026-4806.23.08466-5
115. Brown, J, Yazdi, F, Jodari-Karimi, M, Owen, JG, and Reisin, E. Obstructive sleep apnea and hypertension: updates to a critical relationship. Curr Hypertens Rep . (2022) 24:173–84. doi: 10.1007/s11906-022-01181-w
116. Chaudhary, SC, Gupta, P, Sawlani, KK, Gupta, KK, Singh, A, Usman, K, et al. Obstructive sleep apnea in hypertension. Cureus . (2023) 15:e38229. doi: 10.7759/cureus.38229
117. Mansukhani, MP, Kolla, BP, and Somers, VK. Hypertension and cognitive decline: implications of obstructive sleep apnea. Front Cardiovasc Med . (2019) 6:96. doi: 10.3389/fcvm.2019.00096
118. Ahmed, AM, Nur, SM, and Xiaochen, Y. Association between obstructive sleep apnea and resistant hypertension: systematic review and meta-analysis. Front Med . (2023) 10:1200952. doi: 10.3389/fmed.2023.1200952
119. Schweitzer, PA, Taranto-Montemurro, L, Ojile, JM, Thein, SG, Drake, CL, Rosenberg, R, et al. The combination of aroxybutynin and atomoxetine in the treatment of obstructive sleep apnea (MARIPOSA): a randomized controlled trial. Am J Respir Crit Care Med . (2023) 208:1316–27. doi: 10.1164/rccm.202306-1036OC
120. Rodrigues, MM, Carvalho, PHA, Gabrielli, MFR, Lopes, RN, Garcia Junior, OA, Pereira Filho, VA, et al. How obesity affects nasal function in obstructive sleep apnea: anatomic and volumetric parameters. Braz J Otorhinolaryngol . (2022) 88:296–302. doi: 10.1016/j.bjorl.2020.06.002
121. Jennum, P, and Borgesen, SE. Intracranial pressure and obstructive sleep apnea. Chest . (1989) 95:279–83. doi: 10.1378/chest.95.2.279
122. Rabbani, CC, Saltagi, MZ, and Nelson, RF. The role of obesity, sleep apnea, and elevated intracranial pressure in spontaneous cerebrospinal fluid leaks. Curr Opin Otolaryngol Head Neck Surg . (2019) 27:349–55. doi: 10.1097/MOO.0000000000000562
123. Salman, LA, Shulman, R, and Cohen, JB. Obstructive sleep apnea, hypertension, and cardiovascular risk: epidemiology, pathophysiology, and management. Curr Cardiol Rep . (2020) 22:6. doi: 10.1007/s11886-020-1257-y
124. Zhao, L, Gao, Y, Xu, W, Li, K, Liu, L, and Fan, L. Factors influencing new-onset hypertension in elderly patients with obstructive sleep apnea: a multicenter cohort study. Clin Transl Sci . (2023) 16:2507–18. doi: 10.1111/cts.13631
125. Kabanovski, A, Chan, A, Shapiro, C, and Margolin, E. Obstructive sleep apnea in men with idiopathic intracranial hypertension: a prospective case-control study. J Neuroophthalmol . (2022) 43:531–4. doi: 10.1097/WNO.0000000000001734
126. Espinosa-Muñoz, E, Ruíz-García, FJ, and Puentes-Zarzuela, C. Simultaneous detection of lower gastrointestinal bleeding and hepatic hemangioma in a scintigraphy study with red blood cells labeled with 99m Tc-stannous pyrophosphate. Rev Gastroenterol Mex . (2020) 85:90–1. doi: 10.1016/j.rgmx.2019.06.005
127. Khan, S, Schoenen, J, and Ashina, M. Sphenopalatine ganglion neuromodulation in migraine: what is the rationale? Cephalalgia . (2014) 34:382–91. doi: 10.1177/0333102413512032
128. Triantafyllidi, H, Arvaniti, C, Palaiodimos, L, Vlachos, S, Schoinas, A, Batistaki, C, et al. Infiltration of the sphenopalatine ganglion decreases blood pressure in newly diagnosed and never treated patients with essential hypertension. Int J Cardiol . (2016) 223:345–51. doi: 10.1016/j.ijcard.2016.08.230
Keywords: hypertension, intracranial pressure, Cushing’s mechanism, glymphatics, sympathetic activity, parasympathetic activity, brain self-protection, brain blood flow resistance
Citation: Phillips WT and Schwartz JG (2024) Nasal turbinate lymphatic obstruction: a proposed new paradigm in the etiology of essential hypertension. Front. Med . 11:1380632. doi: 10.3389/fmed.2024.1380632
Received: 01 February 2024; Accepted: 07 August 2024; Published: 16 August 2024.
Reviewed by:
Copyright © 2024 Phillips and Schwartz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY) . The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: William Thomas Phillips, [email protected]
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Thank you for visiting nature.com. You are using a browser version with limited support for CSS. To obtain the best experience, we recommend you use a more up to date browser (or turn off compatibility mode in Internet Explorer). In the meantime, to ensure continued support, we are displaying the site without styles and JavaScript.
- View all journals
- Explore content
- About the journal
- Publish with us
- Sign up for alerts
- Published: 04 May 2020
Female-biased gene flow between two species of Darwin’s finches
- Sangeet Lamichhaney ORCID: orcid.org/0000-0003-4826-0349 1 na1 nAff5 ,
- Fan Han 1 na1 ,
- Matthew T. Webster ORCID: orcid.org/0000-0003-1141-2863 1 ,
- B. Rosemary Grant 2 ,
- Peter R. Grant 2 &
- Leif Andersson ORCID: orcid.org/0000-0002-4085-6968 1 , 3 , 4
Nature Ecology & Evolution volume 4 , pages 979–986 ( 2020 ) Cite this article
2676 Accesses
19 Citations
109 Altmetric
Metrics details
- Ecological genetics
- Evolutionary ecology
The mosaic nature of hybrid genomes is well recognized, but little is known of how they are shaped initially by patterns of breeding, selection, recombination and differential incompatibilities. On the small Galápagos island of Daphne Major, two species of Darwin’s finches, Geospiza fortis and G. scandens , hybridize rarely and back-cross bidirectionally with little or no loss of fitness under conditions of plentiful food. We used whole-genome sequences to compare genomes from periods before and after successful interbreeding followed by back-crossing. We inferred extensive introgression from G. fortis to G. scandens on autosomes and mitochondria but not on the Z chromosome. The unique combination of long-term field observations and genomic data shows that the reduction of gene flow for Z-linked loci primarily reflects female-biased gene flow, arising from a hybrid-male disadvantage in competition for high-quality territories and mates, rather than from genetic incompatibilities at Z-linked loci.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
24,99 € / 30 days
cancel any time
Subscribe to this journal
Receive 12 digital issues and online access to articles
111,21 € per year
only 9,27 € per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout
Similar content being viewed by others

Fine-scale contemporary recombination variation and its fitness consequences in adaptively diverging stickleback fish

Capture and return of sexual genomes by hybridogenetic frogs provides clonal genome enrichment in a sexual species

The genomics and evolution of inter-sexual mimicry and female-limited polymorphisms in damselflies
Data availability.
The Illumina reads have been submitted to the short reads archive ( http://www.ncbi.nlm.nih.gov/sra ) under accession number PRJNA530015 . The following figures have associated raw data: Fig. 1a,b and Extended Data Fig. 1 . The raw data are available in Supplementary Table 1 .
Code availability
The analyses of the data were carried out with publicly available software, and all are cited in the Methods . The custom scripts used are available at https://github.com/sangeet2019/Darwins-Finches .
Mayr, E. Animal Species and Evolution (Harvard Univ. Press, 1963).
Abbott, R. et al. Hybridization and speciation. J. Evol. Biol. 26 , 229–246 (2013).
CAS PubMed Google Scholar
Suarez-Gonzalez, A., Lexer, C. & Cronk, Q. C. B. Adaptive introgression: a plant perspective. Biol. Lett. 14 , 20170688 (2018).
PubMed PubMed Central Google Scholar
Taylor, S. A. & Larson, E. L. Insights from genomes into the evolutionary importance and prevalence of hybridization in nature. Nat. Ecol. Evol. 3 , 170–177 (2019).
PubMed Google Scholar
Edwards, S. V., Potter, S., Schmitt, C. J., Bragg, J. G. & Moritz, C. Reticulation, divergence, and the phylogeography–phylogenetics continuum. Proc. Natl Acad. Sci. USA 113 , 8025–8032 (2016).
CAS PubMed PubMed Central Google Scholar
Elgvin, T. O. et al. The genomic mosaicism of hybrid speciation. Sci. Adv. 3 , e1602996 (2017).
vonHoldt, B. M. et al. Whole-genome sequence analysis shows that two endemic species of North American wolf are admixtures of the coyote and gray wolf. Sci. Adv. 2 , e1501714 (2016).
Larsen, P. A., Marchan-Rivadeneira, M. R. & Baker, R. J. Natural hybridization generates mammalian lineage with species characteristics. Proc. Natl Acad. Sci. USA 107 , 11447–11452 (2010).
Carling, M. D., Lovette, I. J. & Brumfield, R. T. Historical divergence and gene flow: coalescent analyses of mitochondrial, autosomal and sex-linked loci in Passerina buntings. Evolution 64 , 1762–1772 (2010).
Payseur, B. A. & Rieseberg, L. H. A genomic perspective on hybridization and speciation. Mol. Ecol. 25 , 2337–2360 (2016).
Jones, M. R. et al. Adaptive introgression underlies polymorphic seasonal camouflage in snowshoe hares. Science 360 , 1355–1358 (2018).
Liu, K. J. et al. Interspecific introgressive origin of genomic diversity in the house mouse. Proc. Natl Acad. Sci. USA 112 , 196–201 (2015).
Fontaine, M. C. et al. Extensive introgression in a malaria vector species complex revealed by phylogenomics. Science 347 , 1258524 (2015).
Arnold, M. L. & Kunte, K. Adaptive genetic exchange: a tangled history of admixture and evolutionary innovation. Trends Ecol. Evol. 32 , 601–611 (2017).
Pereira, R. J., Barreto, F. S. & Burton, R. S. Ecological novelty by hybridization: experimental evidence for increased thermal tolerance by transgressive segregation in Tigriopus californicus . Evolution 68 , 204–215 (2014).
Hedrick, P. W. Adaptive introgression in animals: examples and comparison to new mutation and standing variation as sources of adaptive variation. Mol. Ecol. 22 , 4606–4618 (2013).
Lewontin, R. C. & Birch, L. C. Hybridization as a source of variation for adaptation to new environments. Evolution 20 , 315–336 (1966).
Campbell, C. R., Poelstra, J. W. & Yoder, A. D. What is speciation genomics? The roles of ecology, gene flow, and genomic architecture in the formation of species. Biol. J. Linn. Soc. 124 , 561–583 (2018).
Google Scholar
Heliconius Genome Consortium. Butterfly genome reveals promiscuous exchange of mimicry adaptations among species. Nature 487 , 94–98 (2012).
Schumer, M., Rosenthal, G. G. & Andolfatto, P. How common is homoploid hybrid speciation? Evolution 68 , 1553–1560 (2014).
Meier, J. I. et al. Ancient hybridization fuels rapid cichlid fish adaptive radiations. Nat. Commun. 8 , 14363 (2017).
Barrera-Guzman, A. O., Aleixo, A., Shawkey, M. D. & Weir, J. T. Hybrid speciation leads to novel male secondary sexual ornamentation of an Amazonian bird. Proc. Natl Acad. Sci. USA 115 , E218–E225 (2018).
Barrera-Guzmán, A. O., Aleixo, A., Shawkey, M. D. & Weir, J. T. Reply to Rosenthal et al.: both premating and postmating isolation likely contributed to manakin hybrid speciation. Proc. Natl Acad. Sci. USA 115 , E4146–E4147 (2018).
Burns, K. J. et al. Phylogenetics and diversification of tanagers (Passeriformes: Thraupidae), the largest radiation of Neotropical songbirds. Mol. Phylogenet. Evol. 75 , 41–77 (2014).
Lamichhaney, S. et al. Evolution of Darwin’s finches and their beaks revealed by genome sequencing. Nature 518 , 371–375 (2015).
Sato, A. et al. Phylogeny of Darwin’s finches as revealed by mtDNA sequences. Proc. Natl Acad. Sci. USA 96 , 5101–5106 (1999).
Peters, K. J., Myers, S. A., Dudaniec, R. Y., O’Connor, J. A. & Kleindorfer, S. Females drive asymmetrical introgression from rare to common species in Darwin’s tree finches. J. Evol. Biol. 30 , 1940–1952 (2017).
Grant, P. R. Ecology and Evolution of Darwin’s Finches (Princeton Univ. Press, 1999).
Grant, P. R. & Grant, B. R. 40 Years of Evolution: Darwin’s Finches on Daphne Major Island (Princeton Univ. Press, 2014).
Grant, P. R. & Grant, B. R. Conspecific versus heterospecific gene exchange between populations of Darwin’s finches. Phil. Trans. R. Soc. B 365 , 1065–1076 (2010).
Grant, B. R. & Grant, P. R. Evolution of Darwin’s finches caused by a rare climatic event. Proc. R. Soc. Lond. B 251 , 111–117 (1993).
Grant, P. R. & Grant, B. R. Hybridization of bird species. Science 256 , 193–197 (1992).
Stemshorn, K. C., Reed, F. A., Nolte, A. W. & Tautz, D. Rapid formation of distinct hybrid lineages after secondary contact of two fish species ( Cottus sp.). Mol. Ecol. 20 , 1475–1491 (2011).
Mallet, J., Besansky, N. & Hahn, M. W. How reticulated are species? Bioessays 38 , 140–149 (2016).
Kearns, A. M. et al. Genomic evidence of speciation reversal in ravens. Nat. Commun. 9 , 906 (2018).
Joseph, L., Drew, A., Mason, I. J. & Peters, J. L. Introgression between non-sister species of honeyeaters (Aves: Meliphagidae) several million years after speciation. Biol. J. Linn. Soc. 128 , 583–591 (2019).
Zhang, G., Parker, P., Li, B., Li, H. & Wang, J. The genome of Darwin’s finch ( Geospiza fortis ). GigaScience https://doi.org/10.5524/100040 (2012).
Baack, E. J. & Rieseberg, L. H. A genomic view of introgression and hybrid speciation. Curr. Opin. Genet. Dev. 17 , 513–518 (2007).
Rheindt, F. E. & Edwards, S. V. Genetic introgression: an integral but neglected component of speciation in birds. Auk 128 , 620–632 (2011).
Lamichhaney, S. et al. A beak size locus in Darwin’s finches facilitated character displacement during a drought. Science 352 , 470–474 (2016).
Grant, P. R. & Grant, B. R. Evolution of character displacement in Darwin’s finches. Science 313 , 224–226 (2006).
Lamichhaney, S. et al. Rapid hybrid speciation in Darwin’s finches. Science 359 , 224–228 (2018).
Coyne, J. A. & Orr, A. R. Speciation (Sinauer, 2004).
Kleindorfer, S. et al. Species collapse via hybridization in Darwin’s tree finches. Am. Nat. 183 , 325–341 (2014).
Hasselman, D. J. et al. Human disturbance causes the formation of a hybrid swarm between two naturally sympatric fish species. Mol. Ecol. 23 , 1137–1152 (2014).
Behm, J. E., Ives, A. R. & Boughman, J. W. Breakdown in postmating isolation and the collapse of a species pair through hybridization. Am. Nat. 175 , 11–26 (2010).
Vonlanthen, P. et al. Eutrophication causes speciation reversal in whitefish adaptive radiations. Nature 482 , 357–362 (2012).
Taylor, E. B. et al. Speciation in reverse: morphological and genetic evidence of the collapse of a three-spined stickleback ( Gasterosteus aculeatus ) species pair. Mol. Ecol. 15 , 343–355 (2006).
Dutheil, J. Y., Munch, K., Nam, K., Mailund, T. & Schierup, M. H. Strong selective sweeps on the X chromosome in the human–chimpanzee ancestor explain its low divergence. PLoS Genet. 11 , e1005451 (2015).
Irwin, D. E. Sex chromosomes and speciation in birds and other ZW systems. Mol. Ecol. 27 , 3831–3851 (2018).
Runemark, A., Eroukhmanoff, F., Nava-Bolanos, A., Hermansen, J. S. & Meier, J. I. Hybridization, sex-specific genomic architecture and local adaptation. Phil. Trans. R. Soc. B 373 , 20170419 (2018).
Lavretsky, P. et al. Speciation genomics and a role for the Z chromosome in the early stages of divergence between Mexican ducks and mallards. Mol. Ecol. 24 , 5364–5378 (2015).
Storchova, R., Reif, J. & Nachman, M. W. Female heterogamety and speciation: reduced introgression of the Z chromosome between two species of nightingales. Evolution 64 , 456–471 (2010).
Hooper, D. M., Griffith, S. C. & Price, T. D. Sex chromosome inversions enforce reproductive isolation across an avian hybrid zone. Mol. Ecol. 28 , 1246–1262 (2019).
Grant, P. R. & Grant, B. R. Adult sex ratio influences mate choice in Darwin’s finches. Proc. Natl Acad. Sci. USA 116 , 12373–12382 (2019).
Grant, P. R. & Grant, B. R. Demography and the genetically effective sizes of two populations of Darwin’s finches. Ecology 73 , 766–784 (1992).
Seehausen, O., Takimoto, G., Roy, D. & Jokela, J. Speciation reversal and biodiversity dynamics with hybridization in changing environments. Mol. Ecol. 17 , 30–44 (2008).
Rudman, S. M. & Schluter, D. Ecological impacts of reverse speciation in threespine stickleback. Curr. Biol. 26 , 490–495 (2016).
Wirtz, P. Mother species–father species: unidirectional hybridization in animals with female choice. Anim. Behav. 58 , 1–12 (1999).
Grant, P. R. & Grant, B. R. Phenotypic and genetic effects of hybridization in Darwin’s finches. Evolution 48 , 297–316 (1994).
Grant, P. R. & Grant, B. R. Role of sexual imprinting in assortative mating and premating isolation in Darwin’s finches. Proc. Natl Acad. Sci. USA 115 , E10879–E10887 (2018).
Ellegren, H. The evolutionary genomics of birds. Annu. Rev. Ecol. Evol. Syst. 44 , 239–259 (2013).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30 , 2114–2120 (2014).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25 , 1754–1760 (2009).
Van der Auwera, G. A. et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinformatics 43 , 10–33 (2013).
Zhang, G. et al. Comparative genomics reveals insights into avian genome evolution and adaptation. Science 346 , 1311–1320 (2014).
Felsenstein, J. PHYLIP—Phylogeny Inference Package (version 3.2). Cladistics 5 , 164–166 (1989).
Schlötterer, C., Tobler, R., Kofler, R. & Nolte, V. Sequencing pools of individuals—mining genome-wide polymorphism data without big funding. Nat. Rev. Genet. 15 , 749–763 (2014).
Browning, B. L., Zhou, Y. & Browning, S. R. A one-penny imputed genome from next-generation reference panels. Am. J. Hum. Genet. 103 , 338–348 (2018).
Grant, P. R. & Grant, B. R. How and Why Species Multiply: The Radiation of Darwin’s Finches (Princeton Univ. Press, 2008).
Download references
Acknowledgements
We thank U. Gustafson for expert wet laboratory assistance and E. Enbody for helpful discussion on the manuscript. The collection of the material, funded by the National Science Foundation (NSF), was conducted with annual permits from the Galápagos National Parks Directorate, with the approval of Princeton University’s Animal Care Committee and in accordance with its protocols, and supported logistically by the Charles Darwin Research Station in Galápagos. The project was supported by Vetenskapsrådet and Knut and Alice Wallenberg Foundation. The genome sequencing was performed by the SNP&SEQ Technology Platform, supported by Uppsala University and SciLifeLab. Computer resources for the bioinformatics analysis were supplied by the Uppsala Multidisciplinary Center for Advanced Computational Science (UPPMAX).
Author information
- Sangeet Lamichhaney
Present address: Department of Biological Sciences, Kent State University, Kent, OH, USA
These authors contributed equally: Sangeet Lamichhaney, Fan Han.
Authors and Affiliations
Science for Life Laboratory, Department of Medical Biochemistry and Microbiology, Uppsala University, Uppsala, Sweden
Sangeet Lamichhaney, Fan Han, Matthew T. Webster & Leif Andersson
Department of Ecology and Evolutionary Biology, Princeton University, Princeton, NJ, USA
B. Rosemary Grant & Peter R. Grant
Department of Animal Breeding and Genetics, Swedish University of Agricultural Sciences, Uppsala, Sweden
Leif Andersson
Department of Veterinary Integrative Biosciences, Texas A&M University, College Station, TX, USA
You can also search for this author in PubMed Google Scholar
Contributions
P.R.G. and B.R.G. collected the material. L.A., P.R.G. and B.R.G. conceived the study. L.A. and M.T.W. led the bioinformatic analysis of the data. S.L. and F.H. performed the bioinformatic analysis and experimental work. L.A., S.L., F.H., B.R.G. and P.R.G. wrote the paper with input from the other authors. All authors approved the manuscript before submission.
Corresponding author
Correspondence to Leif Andersson .
Ethics declarations
Competing interests.
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended data fig. 1.
Beak length and beak depth in six groups of G. scandens and G. fortis population, based on data presented in Supplementary Table 1 .
Extended Data Fig. 2 Beak size (PC1) and beak shape (PC2) of the six groups of finches.
In a Principal Components analysis of beak length, depth and width of all individuals, 30 per group (29 only for early G. fortis ), PC1 explained 64.5% of the variation and PC2 explained an additional 31.4%. The combined groups were heterogeneous in PC1 (F 5,173 = 27.6, P < 0.0001) and PC2 scores (F 5,173 = 497.3, P < 0.0001), more strongly in PC2 (adj R 2 = 0.93) than in PC1 (adj R 2 = 0.43). All pairwise differences in PC2 scores between groups of the same species are statistically significant at P < 0.0001, except for G. scandens early and late pointed groups at P = 0.02. The two groups that contain putatively introgressed individuals, G. fortis late pointed and G. scandens late blunt, do not differ in beak shape (P = 0.72).
Extended Data Fig. 3
Allele frequency of a diagnostic SNP at nucleotide position 16,851 in mtDNA in different groups of G. scandens and G. fortis from Daphne Major.
Extended Data Fig. 4 Normalized genetic distance in four late groups of Darwin’s finches along chromosomes 1, 4 and Z.
Nei’s genetic distance of every 50 kb non-overlapping window was calculated across the genome and only the windows showing relatively high divergence between the early groups (delta genetic distance > 0.15) are presented. Each value was normalized by the difference of the genetic distances between the G. scandens early pointed (SEP) and G. fortis early blunt (FEB) groups.
Extended Data Fig. 5 Density of allele frequencies and their correlation among six groups of Darwin’s finches.
( a ) Density of allele frequency in each group across autosomes and ( b ) on the Z chromosome; the peak density is marked with a dashed blue line. ( c ) Pairwise correlation of allele frequencies among groups on autosomes and ( d ) on the Z chromosome. Correlation coefficients were calculated using Pearson’s correlation test, and all the values were below a significance level of 0.01.
Extended Data Fig. 6
Relative degree of introgression in G. scandens late blunt Delta F ST(SLB) and G. scandens late pointed Delta F ST(SLP) along the genome.
Extended Data Fig. 7
Correlation between two delta F ST measures on each chromosome, and delta F ST(SLB) ≈ delta F ST(SLP) is expected in regions of the genome unaffected by introgression, which is indicated as a red dashed line in each plot together with the Pearson’s correlation coefficient.
Extended Data Fig. 8
Frequency of the ALX1 blunt ( B ) allele in each of the six groups of G. fortis and G. scandens on Daphne Major based on individual genotyping (n = 30 for each pool).
Extended Data Fig. 9
Genotypes for the most significantly differentiated SNPs (n = 6,730) ( F ST > 0.6) from region 2 in Fig. 4a among individually sequenced ground finches ( Geospiza spp .).
Supplementary information
Reporting summary, supplementary table 1.
Morphological data for the G. fortis and G. scandens individuals included in this study.
Rights and permissions
Reprints and permissions
About this article
Cite this article.
Lamichhaney, S., Han, F., Webster, M.T. et al. Female-biased gene flow between two species of Darwin’s finches. Nat Ecol Evol 4 , 979–986 (2020). https://doi.org/10.1038/s41559-020-1183-9
Download citation
Received : 10 April 2019
Accepted : 20 March 2020
Published : 04 May 2020
Issue Date : July 2020
DOI : https://doi.org/10.1038/s41559-020-1183-9
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
This article is cited by
Spatiotemporal variations in retrovirus-host interactions among darwin’s finches.
- Mette Lillie
- Patric Jern
Nature Communications (2022)
Strong bidirectional gene flow between fish lineages separated for over 100,000 years
- Maiko L. Lutz
- Paul Sunnucks
- Alexandra Pavlova
Conservation Genetics (2022)
Exploring potentialities of avian genomic research in Nepalese Himalayas
- Prashant Ghimire
- Nishma Dahal
Avian Research (2021)
Genomic data and multi-species demographic modelling uncover past hybridization between currently allopatric freshwater species
- Sofia L. Mendes
- Miguel P. Machado
- Vitor C. Sousa
Heredity (2021)
Quick links
- Explore articles by subject
- Guide to authors
- Editorial policies
Sign up for the Nature Briefing newsletter — what matters in science, free to your inbox daily.

COMMENTS
Gene flow. Gene flow — also called migration — is any movement of individuals, and/or the genetic material they carry, from one population to another. Gene flow includes lots of different kinds of events, such as pollen being blown to a new destination or people moving to new cities or countries. If genetic variants are carried to a ...
gene flow, the introduction of genetic material (by interbreeding) from one population of a species to another, thereby changing the composition of the gene pool of the receiving population. The introduction of new alleles through gene flow increases variability within the population and makes possible new combinations of traits. In humans gene flow usually comes about through the actual ...
The chart below shows gene flow between the different domains of life. Tree Of Life. A horizontal line shows any place which gene flow allowed genetic variation to pass between the various populations of organisms. It is through this horizontal gene flow that eukaryotes gained the pathways for both mitochondria and plastids such as chloroplasts ...
Gene flow is the transfer of alleles from one population to another population through immigration of individuals.. In population genetics, gene flow (also known as migration and allele flow) is the transfer of genetic material from one population to another. If the rate of gene flow is high enough, then two populations will have equivalent allele frequencies and therefore can be considered a ...
If the assumptions are not met for a gene, the population may evolve for that gene (the gene's allele frequencies may change). Mechanisms of evolution correspond to violations of different Hardy-Weinberg assumptions. They are: mutation, non-random mating, gene flow, finite population size (genetic drift), and natural selection.
Introduction. That gene flow is a fundamental evolutionary force in speciation and adaptation is now widely accepted. Although the role of gene flow was addressed in many theoretical models since the early 1930s (e.g. Haldane 1930; Wright 1931; Slatkin 1987), empirical studies explicitly addressing the role of gene flow began with the advent of genetic markers, which allowed its quantification ...
Hybridization and gene flow are shortcuts to biodiversity that don't always involve differentiation. With human-induced destruction of habitats and climate-change issues pervading the news ...
Genome-wide data hold the key to answering long-standing questions about the mechanisms of speciation, including the role of gene flow. Here, the authors discuss recently developed methods to ...
Natural selection, genetic drift, and gene flow are the mechanisms that cause changes in allele frequencies over time. When one or more of these forces are acting in a population, the population ...
First, speciation may occur by a "budding" process, in that certain populations are removed from gene flow with other populations, which allows them to diverge. If divergence is rapid and gene flow is low, paraphyly may be retained for some time (Funk and Omland 2003). Support for this hypothesis comes from island-specific diversification ...
Gene flow is a key evolutionary process upon which theoretical frameworks of speciation have been primarily founded in sexual organisms. In contrast, bacteria reproduce asexually, and developing a theoretical framework to establish a definition of bacterial species has proven difficult; and some have even questioned the existence of prokaryotic species [1, 2].
Variations in this code, called mutations, lead to unique traits. Gene flow, the mixing of genetic information, maintains consistency within a species. When gene flow is restricted, species can diverge, leading to new species. This process, called speciation, contributes to biodiversity. Created by California Academy of Sciences.
Genetic drift, gene flow, and natural selection may sound similar or even confusing to some. All three are mechanisms in the evolutionary process that have to do with alleles and/or gametes, but there are several significant differences. Discussions about genes and natural selection usually include the term allele. An allele is just one version of a gene found at the same place (locus) on a ...
The gene-for-gene relationship of host-pathogen interaction explained by H. H. Flor in mid of the 20th century set a milestone in understanding the biochemical and genetic basis of plant diseases and several components involved in plant-pathogen interactions. It highlighted the importance of accomplishing differential sets and understanding the pathogen population structure, it further led ...
The swamping hypothesis rests on two key ideas. The first is that gene flow is asymmetric from the center of the range toward the edge, due to decreasing population sizes predicted by the abundant center hypothesis (Figure 1) [9, 12, 13].Although the abundant center hypothesis has been questioned as a general pattern [14, 15], there could still be substantial gene flow from the center of a ...
A somewhat different archaic form, ... time was explained by a balance of gene flow ... expansions rejects the hypothesis of a total replacement. Gene trees based on living humans will only pick ...
Random drift is a more general source of negative linkage disequilibria, and can cause selection for recombination even in large populations, through the chance loss of new favourable mutations. The rate of species-wide substitutions is much too low to drive this mechanism, but local fluctuations in selection, combined with gene flow, may suffice.
Study with Quizlet and memorize flashcards containing terms like The original source of all genetic variation is _____. mutation recombination natural selection sexual reproduction independent assortment, Genetic variation _____. tends to be reduced by when diploid organisms produce gametes is created by the direct action of natural selection must be present in a population before natural ...
A team of scientists from Princeton University and Uppsala University detail their findings of how gene flow between two species of Darwin's finches has affected their beak morphology in the May 4 issue of the journal Nature Ecology and Evolution. Darwin's finches on the Galápagos Islands are an example of a rapid adaptive radiation in ...
Skip to primary navigation; Skip to main content; Skip to primary sidebar; Skip to footer; Image & Use Policy; Translations; UC MUSEUM OF PALEONTOLOGY. Understanding Evolution. Yo
The authors propose that nasal turbinate vasodilation and resultant blood pooling obstruct the flow of cerebrospinal fluid passing through nasal turbinate lymphatics, thereby increasing intracranial pressure. ... This hypothesis explains the rapid worldwide rise in essential hypertension in the last 50 years and offers a novel mechanism and a ...
Gene flow is continuing, and although rare, it occurs between several pairs of species 27, 28. A long-term study of ground finch species on the small island of Daphne Major (34 ha) has documented ...