- Sign In to save searches and organize your favorite content.
- Not registered? Sign up

Recently viewed (0)
- Save Search
- Subscriptions
- Join E-mail List
Patient Case Studies and Panel Discussion: Leukemia – Rare and Emerging Subtypes
- Get Citation Alerts
- Download PDF to Print
Rare and emerging subtypes of leukemia can be incredibly challenging to diagnose and even more challenging to treat. At the NCCN 2019 Annual Congress: Hematologic Malignancies, a panel of experts, moderated by Andrew D. Zelenetz, MD, PhD, were presented with particularly challenging cases in these malignancies and asked to discuss best approaches to treatment.
- Patient Case Study 1
In the first case study, a 77-year-old woman presented with multiple nodular lesions and plaques on her face, chest, and back. She had a history of type 2 diabetes, stage 3 hypertension, hyperlipidemia, coronary heart disease, cerebral infarction, glaucoma, lens extracapsular extraction and posterior chamber intraocular lens implantation, Sjögren syndrome, rheumatoid arthritis, and left axillary vein and brachial vein thrombosis.
She had previously received a conventional therapy of Chinese medicine, but her condition did not improve. Her clinicians performed a bone marrow biopsy and an aspiration biopsy of a nodule on the right side of her face, and immunostaining results revealed the following immunophenotype: CD4+, CD123+, CD43+, CD56+, with Ki-67 level of 30% to 40%.
The patient was diagnosed with blastic plasmacytoid dendritic cell neoplasm, which is a rare blood cancer in the myeloid malignancies family. Andrew D. Zelenetz, MD, PhD, Memorial Sloan Kettering Cancer Center, noted that this disease used to be classified as a variant of acute lymphoblastic leukemia (ALL) and has a distinctive immunophenotype and clinical appearance, characterized by purple skin lesions.
He said a helpful tool for remembering the immunophenotype of this disease is to think “123456”: CD123, CD4, and CD56. Conversely, Nitin Jain, MD, The University of Texas MD Anderson Cancer Center, noted that although this rule of thumb can be helpful, it is important to keep in mind that approximately 10% of patients with this malignancy are actually CD56-negative.
Daniel A. Pollyea, MD, MS, University of Colorado Cancer Center, emphasized the unique phenotypic expression pattern in this malignancy, and the risk of cytopenias due to bone marrow involvement. “Certainly there are patients with bone marrow involvement who don't have cytopenias and have predominant expression of these skin manifestations,” he said. “But I think the CD123 is really the key, because this is a very, very difficult diagnosis to make, and that can be the linchpin.” He added that CD123 expression status is important to know not only for diagnostic purposes but also from a therapeutic perspective. However, many clinical pathologists do not possess the capabilities to test for CD123, so if a diagnosis of blastic plasmacytoid dendritic cell neoplasm is even being entertained, a discussion with a pathologist regarding testing for CD123 is critical.
The nodule on the right side of the patient’s face was surgically excised, and she was treated with gemcitabine, nedaplatin (a second-generation platinum drug used in China that is not approved by the FDA; it is similar to carboplatin and cisplatin), and bleomycin. The patient experienced an initial response to therapy but subsequently developed additional nodular lesions on her arm.
According to Dr. Pollyea, regardless of what transpired with this particular patient, surgical resection of skin lesions did not have a role in this case. “Typically, if the disease is going to respond, the skin lesions are very, very sensitive,” he said. “So there are issues with wound healing if you perform a large resection.”
The panel then discussed tagraxofusp-erzs, a recently approved drug for the treatment of this disorder that has been shown to be highly effective. 1 Dr. Pollyea noted that the mechanism of action of this drug is “quite brilliant.”
“You're taking one of nature's most potent toxins and delivering it directly to a cell population of critical importance in this disease, and potentially the precursor or primitive population of the disease,” he said.
A trial of tagraxofusp treatment in patients with blastic plasmacytoid dendritic cell neoplasms led to durable responses and high complete response rates, particularly in the first-line setting (72%). 1 In relapsed/refractory disease, it was less effective, but “still very effective,” according to Dr. Zelenetz, with a complete response rate of 38%. However, significant toxicity was seen, with capillary leak syndrome a fatal toxicity.
Jae Park, MD, Memorial Sloan Kettering Cancer Center, noted that because of the limited clinical experience with this agent, it is critical to administer the drug in an inpatient setting whenever possible and to closely monitor any patient-related physical changes, including weight fluctuations, kidney function, and respiratory status.
William G. Wierda, MD, PhD, The University of Texas MD Anderson Cancer Center, agreed, adding that he actually treated patients with this compound on a clinical trial before its approval. “During the trial, we were closely monitoring daily weight, albumin, and [liver function], and making daily adjustments in dosing based on what was happening with patients clinically,” he said. “So it's important to be very familiar with the prescribing information.”
Given this particular patient’s age, history, and comorbidities, stem cell transplantation was not an option. However, according to Dr. Park, allotransplant should be considered in these cases whenever possible, and earlier rather than later. “Even with a good response, it becomes difficult to continue this regimen,” he said. “And after [patients] relapse, there are very few treatment options available.”
- Patient Case Study 2
A 28-year-old woman presented with fatigue and lymphadenopathy. Her initial WBC count was 11.1 k/uL with 40% blasts, and she showed hypercellular bone marrow. Her immunophenotype included the following: 88.0% CD45+/–, CD34+, CD19+, CD10+ (variable), CD20– (∼4% of cells stain), sCD22+, CD13–, CD33–, CD38+, CD56–, CD2+/–, CD3–, CD4–, CD8–, CD7–, CD5–, CD117, HLA-DR+, sIg light chain–, cCD79a+, cCD22+, MPO–, cIgM+, and TdT+. After noting the complexity of the patient’s immunophenotype, Dr. Pollyea emphasized the importance of working with a skilled hematopathologist in cases such as this.
The patient was diagnosed with B-cell ALL and treated with the CALGB 10403 regimen. 2 At day 30, bone marrow biopsy showed residual disease with 16% blasts by flow. As her next course of treatment, the patient received blinatumomab for one cycle.
Dr. Jain agreed that this was a reasonable next step, but added that an additional cycle of chemotherapy would also have been feasible. Although the patient was high-risk, he would not yet say treatment had failed after only one treatment cycle.
“I think on the adult side we have to take our cues from the pediatricians who have been so incredibly successful with this disease,” said Dr. Pollyea. “And CALGB 10403 is a regimen that attempts to apply the pediatric regimens to an adolescent/young adult population.” 2
He added that pediatricians tend to stick to protocol, and the protocol for this particular regimen allows for a more extended induction period. “So at this point you should have a lot of concerns about this patient, but I think the protocol allows you to continue.”
About 4 weeks after starting blinatumomab, the patient experienced complete remission confirmed by bone marrow biopsy. She also received 6 cycles of intrathecal chemotherapy throughout the course of her treatment and showed no evidence of central nervous system involvement.
A month later, she presented with enlarged lymph nodes in her groin and neck, and bone marrow biopsy confirmed 63% blasts with an ALL phenotype. A same-day inguinal lymph node biopsy was consistent with lymphoblastic leukemia involvement.
Although the patient experienced a complete remission initially, Dr. Park noted that minimal residual disease (MRD) status was never confirmed. This factor is critical in assessing a patient’s depth of remission, and MRD-positive patients should receive additional therapy sooner rather than later to get to MRD-negative status, he said.
Dr. Jain said that additional diagnostic testing in the form of RNA sequencing would be appropriate in this case, but noted a caveat of the limited availability of this type of testing. The patient underwent next-generation sequencing (NGS), which revealed the following: DIAPH1-PDGFRB fusion; CDKN2A/B - p14 ARF loss exon 1 and CDKN2b loss; PIK3R1 splice site 1746-2A>6; and TP53 N288fs*60.
According to Dr. Park, interpreting NGS data can be difficult, and misinterpretation can lead to the wrong choice of treatment. This again underlines the importance of consulting with a skilled pathologist or other experienced ALL expert to assist in interpreting mutation profiles.
The patient was determined to have Ph-like ALL (a newly recognized entity of Ph-negative ALL with a poor prognosis) and was enrolled in the KTE-CA19 CAR-T (axicabtagene ciloleucel [axi-cel]) trial ( ClinicalTrials.gov identifier: NCT02614066). She received cytoreductive chemotherapy with hyperCVAD part A before apheresis for CAR-T generation, and experienced favorable cytoreduction (she received fludarabine/cyclophosphamide for lymphodepletion). She then received a post–CAR-T infusion and showed no response; her blast count increased from 0.42 to 80.35 within a week.
“This is just a tough case,” said Dr. Park, noting the unusually refractory nature of the disease. “Initial response rates to CAR-T cell therapy are approximately 80%, so she’s already in the very unlucky 20% of cases,” he said.
Dr. Jain described 2 subtypes of Ph-like ALL: approximately half are CRLF2 -rearranged, 3 and these patients should ideally be referred to a clinical trial. The other half are nonrearranged, 3 and these patients should be referred for RNA sequencing to determine fusion genes.
No response was seen to further treatment, and the patient chose to continue care in hospice.
According to Dr. Zelenetz, incorporation of comprehensive genetic analysis and fluorescence in situ hybridization testing is important to identify high-risk patients (such as those with Ph-like phenotype) and plan for allogeneic hematopoietic stem cell transplantation (alloHSCT) or referral to clinical trials as early as possible.
MRD assessment by flow and/or NGS is critical to assess depth of response, modification of therapy, and candidacy for early alloHSCT. Dr. Park noted that both gene sequencing tests are validated, so patient preference should take priority.
Incorporation of tyrosine kinase inhibitors (TKIs) in Ph-like ALL is being investigated in clinical trials, and patients with this disease should be referred earlier rather than later, added Dr. Zelenetz. “But the nuance to that is understanding how to integrate TKIs into this entity, which is going to be dependent on understanding the mechanisms involved in the disease,” he said. “It won’t be just one TKI [that everyone receives]; it's much more complicated than that, unfortunately.”
Dr. Jain added that although Ph-like ALL has been established as high risk in the setting of chemotherapy, its classification remains to be determined in the new era of targeted therapies. “Some emerging data suggest that blinatumomab, inotuzumab, and CAR-T-cell therapy may overcome the negative prognostication of Ph-like ALL,” he said. “So those are some data we’ll hopefully see at the ASH Annual Meeting.”
Jarrod Holmes, MD, Annadel Medical Group, also participated in the panel discussion.
Pemmaraju N , Lane AA , Sweet KL , et al. . Tagraxofusp in blastic plasmacytoid dendritic-cell neoplasm . N Engl J Med 2019 ; 380 : 1628 – 1637 .
- Search Google Scholar
- Export Citation
Stock W , Luger SW , Advani AS , et al. . A pediatric regimen for older adolescents and young adults with acute lymphoblastic leukemia: results of CALGB 10403 . Blood 2016 ; 133 : 1548 – 1559 .
Jain N , Roberts KG , Jabbour E , et al. . Ph-like acute lymphoblastic leukemia: a high-risk subtype in adults . Blood 2017 ; 129 : 572 – 581 .
Disclosures: Dr. Zelenetz has disclosed that he receives research support from Genentech/Roche, Gilead, MEI, and BeiGene; he has been a consultant for Celegene/JUNO, Genentech/Roche, Gilead, BeiGene, Pharmacyclics, Jansen, Amgen, Astra‐Zeneca, Novartis, and MEI Pharma; and he is on the Scientific Advisory Board of the Lymphoma Research Foundation and Adaptive Biotechnologies. Dr. Jain has disclosed that he is a consultant for AbbVie, Inc., AstraZeneca Pharmaceuticals LP, Genentech, Inc., Janssen Pharmaceutica Products, LP, Adaptive Biotechnologies, Precision Biosciences, Verastem, and Pharmacyclics; receives grant/research support from AbbVie, Inc., AstraZeneca Pharmaceuticals LP, Bristol-Myers Squibb Company, Genentech, Inc., Incyte Corporation, Adaptive Biotechnologies, ADC Therapeutics, Cellectis, Precision Biosciences, Servier, Verastem, Pfizer, Inc., and Pharmacyclics; is a scientific advisor for AbbVie, Inc., AstraZeneca Pharmaceuticals LP, Genentech, Inc., Janssen Pharmaceutica Products, LP, Adaptive Biotechnologies, Precision Biosciences, Verastem, and Pharmacyclics; and has received honoraria from AbbVie, Inc., AstraZeneca Pharmaceuticals LP, Genentech, Inc., Janssen Pharmaceutica Products, LP, Adaptive Biotechnologies, Precision Biosciences, Verastem, and Pharmacyclics. Dr. Park has disclosed that he receives grant/research support from Amgen Inc., Genentech, Inc., Incyte Corporation, Juno Therapeutics, Inc., Kite Pharma, Novartis Pharmaceuticals Corporation, and Servier; and is a scientific advisor for from Amgen Inc., AstraZeneca Pharmaceuticals LP, GlaxoSmithKline, Incyte Corporation, Kite Pharma, Novartis Pharmaceuticals Corporation, Allogene Therapeutics, Autolus Therapeutics plc, and Takeda Pharmaceuticals North America, Inc. Dr. Pollyea has disclosed that he is a scientific advisor for AbbVie, Inc., Agios, Inc., Celgene Corporation, Daiichi-Sankyo Co., Forty Seven, Inc., Janssen Pharmaceutica Products, LP, Pfizer Inc., and Takeda Pharmaceuticals North America, Inc. Dr. Wierda has disclosed that he is a consultant for Genzyme Corporation and receives grant/research support from AbbVie, Inc., Acerta Pharma, Genentech, Inc., Gilead Sciences, Inc., Janssen Pharmaceutica Products, LP, Juno Therapeutics, Inc., Karyopharm Therapeutics, Kite Pharma, Cyclacel Pharmaceuticals, Inc., GlaxoSmithKline/Novartis Pharmaceuticals Corporation, Loxo Oncology, Inc., miRagen Therapeutics, Inc., Oncternal Therapeutics, Inc., Xencor, Inc., Pharmacyclics, and Sunesis Pharmaceuticals, Inc. Dr. Holmes has disclosed that he has no financial interests, arrangements, affiliations, or commercial interests with the manufacturers of any products discussed in this article or their competitors.
Journal of the National Comprehensive Cancer Network
Article Sections
Article information.
- Get Permissions
- Similar articles in PubMed
Google Scholar
Related articles.
| All Time | Past Year | Past 30 Days | |
|---|---|---|---|
| Abstract Views | 0 | 0 | 0 |
| Full Text Views | 5105 | 1041 | 40 |
| PDF Downloads | 3620 | 670 | 29 |
| EPUB Downloads | 0 | 0 | 0 |
- Advertising
- Terms of Use
- Privacy Policy
- Permissions
© 2019-2024 National Comprehensive Cancer Network
Powered by:
- [81.177.180.204]
- 81.177.180.204
Character limit 500 /500
- Case report
- Open access
- Published: 05 February 2022
A case report of pediatric acute lymphoblastic leukemia with e8a2 BCR/ABL1 fusion transcript
- Aleksandra Mroczkowska ORCID: orcid.org/0000-0002-8837-6517 1 ,
- Bożena Jaźwiec 1 , 2 ,
- Justyna Urbańska-Rakus 3 ,
- Sylwia Szymanowska 1 ,
- Anna Tessmann 1 ,
- Sonia Pająk 3 ,
- Katarzyna Machnik 3 ,
- Olga Haus 4 &
- Tomasz Wróbel 2
BMC Medical Genomics volume 15 , Article number: 20 ( 2022 ) Cite this article
9205 Accesses
4 Citations
Metrics details
Acute lymphoblastic leukemia is the most common type of cancer in children. Most often it affects the age group between 2 and 5 years of age. Studies have shown an improvement in general survivability, more than 90% 5-year overall survival (OS). Current treatment protocols for acute lymphoblastic leukemia require verification of the presence of favorable and unfavorable genetic abnormalities, which help qualify patients to the appropriate risk group and select a more suitable treatment. The presence of the BCR/ABL1 fusion gene stratifies the patient into a high-risk group and requires special treatment with tyrosine kinase inhibitors (TKI). The three dominant mRNA transcripts are e1a2, e13a2, and e14a2. Nevertheless, cases of atypical BCR/ABL1 transcripts have also been reported.
Case presentation
This paper presents the case of a pediatric patient with Ph + B-cell precursor acute lymphoblastic leukemia with rare atypical e8a2 BCR/ABL1 fusion transcript. Our patient achieved complete remission after 33 days of treatment. Molecular and cytogenetic studies in TP1 did not reveal the presence of the BCR/ABL1 transcript. The PCR-MRD test in TP1b was negative, the patient did not require hematopoietic stem cell transplantation.
Genetic evaluation of the bone marrow sample is crucial in the initial stage of the diagnosis. Fluorescent in situ hybridization and reverse transcriptase polymerase chain reaction with Sanger sequencing are the appropriate methods used in the detection of rare variants of BCR/ABL1 transcripts.
Peer Review reports
Acute lymphoblastic leukemia (ALL) is the most common childhood malignancy. ALL is a heterogeneous neoplasm derived from the precursors of the lymphoid lineage. About 80–85% of cases are B-cell precursor leukemias, while T-lineage leukemias are about 15–20%. The ALL diagnoses are based on certain criteria including clinical presentation, laboratory tests, a bone marrow biopsy, immunophenotypic analysis and genetic tests. Currently, cytogenetic and molecular tests play a very important role in determining prognosis and stratification for suitable treatment of pediatric ALL [ 1 , 2 ]. The typical recurrent translocations occurring in ALL are t(12;21)(p13;q22) causing ETV6/RUNX1 , t(1;19)(q23;p13) causing TCF3/PBX1 , t(9;22)(q34;q11.2) causing BCR/ABL1 , and the most common rearrangement of KMT2A gene, t(4;11)(q21;q23) causing KMT2A/AFF1 . ETV6/RUNX1 is associated with a favorable prognosis and the last three genetic abnormalities have unfavorable outcomes [ 3 , 4 ].
BCR/ABL1 fusion transcripts occur approximately in 2–5% cases of childhood ALL and the frequency of BCR/ABL1(+)ALL increases with the patient’s age [ 5 ]. ALL cases with this genetic abnormality are associated with poor outcome and are qualified to the high risk group. Due to the introduction of tyrosine kinase inhibitors to the therapy, the prognosis of Ph + patients has improved.
The most common mRNA transcripts of BCR/ABL1: e1a2, e13a2, e14a2, occur in about 99% of Ph + cases. Approximately 70% of Ph + ALL patients have an e1a2 transcript and more than 25% e13a2 or e14a2. 1% of patients with Ph + shows atypical transcripts like e19a2, e6a2, e1a3, e13a3, e14a3 and e8a2 [ 6 ].
We present here a case of a pediatric patient with Ph + BCP-ALL (B cell precursor ALL) with an e8a2 BCR/ABL1 transcript.
An 11-year-old boy was admitted to the Unit of Pediatric Hematology and Oncology, City Hospital, Chorzów, Poland due to a suspicion of acute leukemia. Five days before admission to the hospital, he developed a severe and difficult to stop nosebleed. Since then, the boy was experienced weakness, lethargy, lack of appetite. Additionally he developed abdominal pain, a headache and nausea. Physical examination revealed pale skin with petechiae, inflammation of the gingiva, tooth decay and splenomegaly. Lymphadenopathy, hepatomegaly and the presence of a Central Nervous System (CNS) disease/leukemia were not observed. Family Health History has no indication of any genetic, hematologic or cancerous diseases. Patient was not exposed to any physical (i.e. ionic radiation) or chemical factors (organic solvents, pesticides, herbicides, paints, lacquers) during childhood nor fetal period. He was born out of second pregnancy, first childbirth (first pregnancy ended due to spontaneous miscarriage around eighth week). Weight at birth 2400 g. Mother's age at birth: 19, father: 21. Patient has younger step-sister (same mother, different father), showing no symptoms of ALL or any other hematological disorders.
By the time of diagnosis of ALL, the patient had been sick sporadically and had no routine blood tests—including morphology. The patient has not taken any medications on a permanent basis.
The laboratory results showed: white blood cell 206,900/µl, platelet count 142,000/µl and hemoglobin level 10.2 g/dl. The bone marrow was highly cellular, represented by a homogeneous population of small blasts with lymphoid morphology (88.5%). Flow cytometric analysis showed BCP-ALL phenotype: CD45dim + , CD38 + , CD34(+), CD81(+), CD24(+), CD19(+), CD79a(+), TdT(+), CD10(+), CDdim33(+), CD20dim(+), CD22dim(+), CD15(-), CD117(-). The boy was diagnosed with common B-cell precursor ALL and qualified for treatment according to the AIEOP-BFM ALL 2017 protocol.
The cytogenetic and molecular examinations of the patient's bone marrow were performed by the Laboratory of Molecular Biology and Cytogenetics at the University Clinical Hospital in Wroclaw. Karyotype analysis and fluorescence in situ hybridization (FISH) was performed on the bone marrow sample according to the AIEOP-BFM ALL 2017 protocol. According to the protocol, tests for genetic diagnostics were performed. By day 6, a FISH test was performed to obtain a result for the presence of the Philadelphia chromosome. Up to day 33, the FISH test was performed for the frequent genetic aberrations: ETV6/RUNX1 translocation and rearrangements in the KMT2A and TCF3 genes. At the same time, molecular tests were carried out using the RT-PCR method for the presence of the BCR/ABL1 and KMT2A/AFF1 fusion gene. G-banded chromosome analysis revealed an abnormal male karyotype 46,XY,t(9;22)(q34;q11) [ 11 ]/46,XY [ 9 ] (Fig. 1 A). The FISH study showed no rearrangements in ETV6/RUNX1, TCF3 (MetaSystems Probes, Germany) or KMT2A (Vysis, Abbott Molecular, Illinois, USA). The FISH study performed with the BCR/ABL1 dual color, dual fusion translocation probe (Vysis, Abbott Molecular, Illinois, USA) disclosed a typical translocation pattern 2 green/orange BCR/ABL1 fusion signals, 1 green BCR signal, and one orange ABL1 signal in 90% of the interphase cells (Fig. 1 B). Reverse transcription-polymerase chain reaction (RT-PCR) was performed to detect the presence or absence of the KMT2A /AFF1 and BCR/ABL1 fusion gene using primers as per JJM van Dongen et al. [ 7 ]. The test was negative in both cases. Due to the positive result of the FISH test for BCR/ABL1 , another RT-PCR was performed in order to search for atypical BCR/ABL1 transcripts. New RT-PCR analysis was performed based on primers BCR-6 and ABL-3 published by T. Burmeister and R. Reinhardt [ 6 ]. Electrophoresis showed a band of ~ 489 bp (Fig. 2 A). Sanger sequencing confirmed the direct junction between exon 8 of BCR (NM_004327.4) and exon 2 of ABL1 (NM_005157.6) (Fig. 2 B). The Sanger sequencing was important because this method determined the type of transcript by analyzing the direct junction between exons. Transcript type information is crucial for monitoring the presence of BCR/ABL1 transcript by RT-PCR method.

A —Conventional G-banding karyotype analysis showing typical translocation between chromosome 9 and 22. B —FISH analysis on interphase and metaphase with LSI BCR/ABL1 Dual Color, Dual Fusion Translocation Probe

A —Detection of e8a2 BCR/ABL1 transcript by RT-PCR. Lane 1: size marker; lane 2: patient sample; lane 3: negative control, lane 4: internal reference gene—ABL1. B —Sanger sequencing demonstrating the direct junction between BCR exon e8 and ABL1 exon a2
The patient’s induction therapy started according to the protocol IA-Pred. On the 8th day of treatment, the patient had a poor response to prednisone. Due to the presence of the BCR /ABL1 fusion gene, further treatment was performed according to the EsPhALL 2009 protocol (European intergroup study of post-induction treatment of Philadelphia-chromosome-positive ALL). Imatinib at a dose of 300 mg/m 2 daily was started on day 15 of treatment, but on day 28 was withheld due to hepatotoxicity (WHO grade III). Evolution of peripheral blood cell counts during therapy is presented in Table 1 . In accordance to protocol, the patient's bone marrow was collected on days 15 and 33 of treatment. Examination of the bone marrow sample on day 15 revealed 15.4% blast cells in bone marrow morphology. Flow cytometry (FCM) revealed 23.48% of blasts. On day 33 (TP1), the bone marrow was already aplastic. Nevertheless, a PCR-MRD (Minimal Residual Disease) result was obtained. MRD in TP1 was low-positive (< 10 −4 ). Bone marrow smear revealed a total of 2.6% of blasts. Despite the poor quality of the material in TP1, it was also possible to perform a FISH study (Fig. 3 A) and RT-PCR test (Fig. 3 B). Both molecular and cytogenetic tests were negative. According to the EsPhALL 2009 protocol the boy should have been classified as poor risk Ph(+) ALL group because of PPR (prednisone poor responder) on the 8th day, but due to complete remission on day 33 (LBL 1.2%, PC-MRD < 10 –4 ) he was classified as good risk Ph(+) ALL group. From about day 32 of treatment, the patient reported abdominal pain, constipation, nausea and vomiting. Physical examinations showed hepatomegaly and lazy intestinal peristalsis. The symptoms were most likely caused by paralytic intestinal obstruction after chemotherapy. Additionally, the patient developed a fungal infection of the bladder.

A —FISH study on day 33 of treatment, B —RT-PCR test on day 33 of treatment. Lane 1: size marker; lane 2: positive control; lane 3: patient; lane 4: negative control, lane 5: internal reference gene—ABL1
Due to the general condition of the patient, consolidation treatment was delayed by 25 days. After this time, according to the EsPhALL 2009 protocol the IB protocol was started and Imatinib was resumed. Another PCR-MRD test was performed on day 17 of treatment (TP1b) of the IB protocol. PCR-MRD in TP1b was negative. Therefore the patient continued chemotherapy without qualification for HSCT (Hematopoietic Stem Cell Transplantation). The patient after consolidation therapy was in haematological remission of ALL. The patient remains without a transplant for 8 months after diagnosis.
Discussion and conclusions
The very rare e8a2 transcript (about 8% from 1% of non-typical BCR/ABL1 transcripts) has been reported mainly in cases of chronic myeloid leukemia (CML) [ 8 , 9 , 10 , 11 , 12 , 13 , 18 ]. Two cases have been reported in adult ALL [ 14 , 15 ]. The e8a2 BCR/ABL1 transcript could be associated with worse prognosis than the e13a2 or the e14a2 transcripts in CML patients. However, there were cases of good response to treatment with imatinib with an achievement of a major molecular response [ 8 , 10 , 12 ]. CML cases with this transcript that have been reported so far, additionally had insertions from ABL1 intron 1b or 1a, from BCR intron 8 or another gene such as PRDM12 , MAST2 [ 11 , 16 , 17 ]. Only one patient with CML and e8a2 BCR/ABL1 transcript had no additional insertions and after treatment with imatinib achieved a complete cytogenetic response [ 12 ]. In adult acute lymphoblastic leukemia one case was reported with insertion of 2 nucleotides from ABL1 intron 1a [ 14 ]. One adult ALL woman that had RALGPS1 exon 8 inserted into the fusion, was treated with FLAG-Ida (fludarabine, cytarabine, granulocyte-colony stimulating factor [G-CSF], idarubicin) and dasatinib and after re-induction therapy achieved hematological, cytogenetic and molecular remission [ 15 ]. Unfortunately, the e8a2 variant in adult ALL patients is so rare, that its impact on outcome remains unknown. To the best of our knowledge, our patient is the first pediatric ALL case with e8a2 BCR/ABL1 transcript. Our case sequencing analysis revealed e8a2 BCR/ABL1 transcript without any insertion. Creation of the e8a2 transcript by the exact fusion of BCR exon e8 to ABL1 exon a2 could encode an oncogenic protein, therefore our patient was qualified for treatment with the EsPhALL 2009 protocol. Our patient achieved complete remission after 33 days of treatment. Molecular and cytogenetic studies in TP1 did not reveal the presence of the BCR/ABL1 transcript. The PCR-MRD test in TP1b was negative, the patient did not require hematopoietic stem cell transplantation.
The presence of the BCR/ABL1 fusion gene is considered an unfavorable genetic abnormality and is associated with poor prognosis but survival has improved with the development of TKI. Our case shows that atypical transcripts of BCR/ABL1 also occur in cases other than CML or adult ALL. RT-PCR and sequencing are appropriate methods for identifying these atypical transcripts. Using both conventional cytogenetics and molecular methods, we are able to detect many genetic changes occurring in leukemias. It is important to identify them accurately and use this information to monitor the patient’s treatments. The monitoring of the presence and quantity of the BCR/ABL1 transcript using the RT-qPCR method is a gold standard in monitoring of Ph + patients with chronic myeloid leukemia. This method can also be used in monitoring of Ph + ALL patients to assess treatment efficiency. For proper patient monitoring it is important to evaluate the type of transcript at the time of diagnosis. Detection of a rare atypical transcript may affect the patient's treatment and may be associated with a worse prognosis.
Availability of data and materials
The Sanger Sequencing data generated in the study has been submitted to NCBI GenBank BankIt with the accession number OL672741; https://www.ncbi.nlm.nih.gov/nuccore/OL672741 . Reference sequences used in this study are available in the following link: https://www.ncbi.nlm.nih.gov/nuccore/NM_004327.4 ; https://www.ncbi.nlm.nih.gov/nuccore/NM_005157.6 . https://www.ncbi.nlm.nih.gov/nuccore/MF925339.1/ .
Abbreviations
Overall survival
Tyrosine kinase inhibitors
B cell precursor Acute Lymphoblastic Leukemia
Central Nervous System
Fluorescence in situ hybridization
Reverse transcription-polymerase chain reaction
Hematopoietic Stem Cell Transplantation
Chronic myeloid leukemia
Granulocyte-colony stimulating factor
Tasian S, Loh M, Hunger S. Childhood acute lymphoblastic leukemia: integrating genomics into therapy. Cancer. 2015;121(20):3577–90. https://doi.org/10.1002/cncr.29573 .
Article PubMed Google Scholar
Moorman A. The clinical relevance of chromosomal and genomic abnormalities in B-cell precursor acute lymphoblastic leukaemia. Blood Rev. 2012;26(3):123–35. https://doi.org/10.1016/j.blre.2012.01.001 .
Article CAS PubMed Google Scholar
Terwilliger T, Abdul-Hay M. Acute lymphoblastic leukemia: a comprehensive review and 2017 update. Blood Cancer J. 2017;7(6): e577. https://doi.org/10.1038/bcj.2017.53 .
Article CAS PubMed PubMed Central Google Scholar
Inaba H, Mullighan CG. Pediatric acute lymphoblastic leukemia. Haematologica. 2020;105(11):2524–39. https://doi.org/10.3324/haematol.2020.247031 .
Mohseni M, Uludag H, Brandwein JM. Advances in biology of acute lymphoblastic leukemia (ALL) and therapeutic implications. Am J Blood Res. 2018;8(4):29–56.
CAS PubMed PubMed Central Google Scholar
Burmeister T, Reinhardt R. A multiplex PCR for improved detection of typical and atypical BCR-ABL fusion transcripts. Leuk Res. 2008;32(4):579–85. https://doi.org/10.1016/j.leukres.2007.08.017 .
Van Dongen JJ, Macintyre EA, Gabert JA, Delabesse E, Rossi V, Saglio G, et al. Standardized RT-PCR analysis of fusion gene transcripts from chromosome aberrations in acute leukemia for detection of minimal residual disease. Report of the BIOMED-1 Concerted Action: investigation of minimal residual disease in acute leukemia. Leukemia. 1999;13(12):1901–28. https://doi.org/10.1038/sj.leu.2401592 .
Cayuela JM, Rousselot P, Nicolini F, Espinouse D, Ollagnier C, Bui-Thi MH, et al. Identification of a rare e8a2 BCR–ABL fusion gene in three novel chronic myeloid leukemia patients treated with imatinib. Leukemia. 2005;19(12):2334–6. https://doi.org/10.1038/sj.leu.2403986 .
Demehri S, Paschka P, Schultheis B, Lange T, Koizumi T, Sugimoto T, et al. e8a2 BCR–ABL: more frequent than other atypical BCR–ABL variants? Leukemia. 2005;19(4):681–4. https://doi.org/10.1038/sj.leu.2403604 .
Tchirkov A, Couderc JL, Perissel B, Goumy C, Regnier A, Uhrham-mer N, et al. Major molecular response to imatinib in a patient with chronic myeloid leukemia expressing a novel form of e8a2 BCR–ABL transcript. Leukemia. 2006;20(1):167–8. https://doi.org/10.1038/sj.leu.2404012 .
Park IJ, Lim YA, Lee WG, Park JS, Kim HC, Lee H-J, Cho SR. A case of chronic myelogenous leukemia with e8a2 fusion transcript. Cancer Genet Cytogenet. 2008;185(2):106–8. https://doi.org/10.1016/j.cancergencyto.2008.06.001 .
Jin C, Zhu X, Xiao M, Liu S, Liu X, Liu J, Xu X, et al. A novel e8a2BCR-ABL1 fusion transcript without insertion sequence in a patient with chronic myeloid leukemia. Ann Lab Med. 2018;38(2):169–71.
Article Google Scholar
Branford S, Rudzki Z, Hughes TP. A novel BCR-ABL transcript (e8a2) with the insertion of an inverted sequence of ABL intron 1b in a patient with Philadelphia-positive chronic myeloid leukaemia. Br J Haematol. 2000;109(3):635–7. https://doi.org/10.1046/j.1365-2141.2000.02042 .
Kim MJ, Yoon HJ, Park TS. The e8a2 fusion transcript in B lymphoblastic leukemia with BCR-ABL1 rearrangement. Korean J Hematol. 2012;47(3):161. https://doi.org/10.5045/kjh.2012.47.3.161 .
Article PubMed PubMed Central Google Scholar
McCarron SL, Kelly J, Coen N, McCabe S, Fay M, O’Dwyer M, et al. A novel e8a2 BCR-ABL1 fusion with insertion of RALGPS1 exon 8 in a patient with relapsed Philadelphia chromosome-positive acute lymphoblastic leukemia. Leuk Lymphoma. 2011;52(5):919–21. https://doi.org/10.3109/10428194.2011.555025 .
Riva E, Manrique Arechavaleta G, De Almeida C, Costa V, Fernandez Del Campo M, Ifran González S, Uriarte R. A novel e8a2 BCR-ABL1 fusion with insertion of MAST2 exon 2 in a four-way translocation t (1;17;9;22) (p35;q24;q44;q11) in a patient with chronic myeloid leukemia. Leuk Lymphoma. 2016;57(1):203–5. https://doi.org/10.3109/10428194.2015.1043549 .
Reid AG, Nacheva EP. A potential role for PRDM12 in the pathogenesis of chronic myeloid leukaemia with derivative chromosome 9 deletion. Leukemia. 2004;18(1):178–80. https://doi.org/10.1038/sj.leu.2403162 .
Qin YZ, Jiang Q, Jiang H, Lai Y-Y, Shi H-X, Chen W-M, et al. Prevalence and outcomes of uncommon BCR/ABL1 fusion transcripts in patients with chronic myeloid leukaemia: data from a single centre. Br J Haematol. 2018;182(5):693–700. https://doi.org/10.1111/bjh.15453 .
Download references
Acknowledgements
Not applicable.
No funding was obtained for this study.
Author information
Authors and affiliations.
Laboratory of Molecular Biology and Cytogenetics, Department of Hematology, Blood Neoplasms and Bone Marrow Transplantation, Wroclaw Medical University, Wrocław, Poland
Aleksandra Mroczkowska, Bożena Jaźwiec, Sylwia Szymanowska & Anna Tessmann
Department of Hematology, Blood Neoplasms and Bone Marrow Transplantation, Wroclaw Medical University, Wrocław, Poland
Bożena Jaźwiec & Tomasz Wróbel
Unit of Pediatric Hematology and Oncology, City Hospital, Chorzow, Poland
Justyna Urbańska-Rakus, Sonia Pająk & Katarzyna Machnik
Department of Clinical Genetics, Faculty of Medicine, Collegium Medicum in Bydgoszcz, Nicolaus Copernicus University in Torun, Bydgoszcz, Poland
You can also search for this author in PubMed Google Scholar
Contributions
AM wrote the manuscript with support from TW and OH. BJ, AM conducted molecular genetics experiments and interpreted the Sanger sequencing data. SS, AT performed cytogenetical experiments. JU-R, SP, KM contributed to the clinical part of the study, prepared a clinical data and edited a clinical part of manuscript. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Correspondence to Aleksandra Mroczkowska .
Ethics declarations
Ethics approval and consent to participate.
This study was approved by the ethics committee of Wroclaw Medical University, Poland (committee’s reference number: KB 716/2018). Written consent to participate was obtained from patient which served as negative control.
Consent for publication
Written informed consent was obtained from the patient’s parents for publication of this case report and any accompanying images. Written consent for publication was also obtained from patient which served as negative control.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's note.
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/ . The Creative Commons Public Domain Dedication waiver ( http://creativecommons.org/publicdomain/zero/1.0/ ) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
Reprints and permissions
About this article
Cite this article.
Mroczkowska, A., Jaźwiec, B., Urbańska-Rakus, J. et al. A case report of pediatric acute lymphoblastic leukemia with e8a2 BCR/ABL1 fusion transcript. BMC Med Genomics 15 , 20 (2022). https://doi.org/10.1186/s12920-022-01169-0
Download citation
Received : 01 July 2021
Accepted : 27 January 2022
Published : 05 February 2022
DOI : https://doi.org/10.1186/s12920-022-01169-0
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
- Acute lymphoblastic leukemia
BMC Medical Genomics
ISSN: 1755-8794
- Submission enquiries: [email protected]
- General enquiries: [email protected]
- Case report
- Open access
- Published: 18 August 2024
Transformation into acute myeloid leukemia with t(8;21)(q22;q22.1); RUNX1::RUNX1T1 from JAK2 -mutated essential thrombocythemia: a case report
- Chie Asou 1 ,
- Tomoyuki Sakamoto 1 ,
- Kodai Suzuki 1 ,
- Itoko Okuda 1 ,
- Atsushi Osaki 1 ,
- Ryohei Abe 1 ,
- Yoshihiro Ito 1 ,
- Emi Kakegawa 1 ,
- Yoshitaka Miyakawa 1 ,
- Yasuhito Terui 1 &
- Yuichi Nakamura ORCID: orcid.org/0000-0001-9518-8453 1
Journal of Medical Case Reports volume 18 , Article number: 372 ( 2024 ) Cite this article
Metrics details
Blast transformation is a rare but well-recognized event in Philadelphia-negative myeloproliferative neoplasms associated with a poor prognosis. Secondary acute myeloid leukemias evolving from myeloproliferative neoplasms are characterized by a unique set of cytogenetic and molecular features distinct from de novo disease. t(8;21) (q22;q22.1); RUNX1::RUNX1T1 , one of the most frequent cytogenetic abnormalities in de novo acute myeloid leukemia, is rarely observed in post-myeloproliferative neoplasm acute myeloid leukemia. Here we report a case of secondary acute myeloid leukemia with t(8;21) evolving from JAK2 -mutated essential thrombocythemia.
Case presentation
The patient was a 74-year-old Japanese woman who was referred because of thrombocytosis (platelets 1046 × 10 9 /L). Bone marrow was hypercellular with increase of megakaryocytes. Chromosomal analysis presented normal karyotype and genetic test revealed JAK2 V617F mutation. She was diagnosed with essential thrombocythemia. Thrombocytosis had been well controlled by oral administration of hydroxyurea; 2 years after the initial diagnosis with ET, she presented with leukocytosis (white blood cells 14.0 × 10 9 /L with 82% of blasts), anemia (hemoglobin 91 g/L), and thrombocytopenia (platelets 24 × 10 9 /L). Bone marrow was hypercellular and filled with 80% of myeloperoxidase-positive blasts bearing Auer rods. Chromosomal analysis revealed t(8;21) (q22;q22.1) and flow cytometry presented positivity of CD 13, 19, 34, and 56. Molecular analysis showed the coexistence of RUNX1::RUNX1T1 chimeric transcript and heterozygous JAK2 V617F mutation in leukemic blasts. She was diagnosed with secondary acute myeloid leukemia with t(8;21)(q22;q22.1); RUNX1::RUNX1T1 evolving from essential thrombocythemia. She was treated with combination chemotherapy with venetoclax and azacytidine. After the first cycle of the therapy, blasts disappeared from peripheral blood and decreased to 1.4% in bone marrow. After the chemotherapy, RUNX1::RUNX1T1 chimeric transcript disappeared, whereas mutation of JAK2 V617F was still present in peripheral leukocytes.
Conclusions
To our best knowledge, the present case is the first one with JAK2 mutation preceding the acquisition of t(8;21). Our result suggests that t(8;21); RUNX1::RUNX1T1 can be generated as a late event in the progression of JAK2 -mutated myeloproliferative neoplasms. The case presented typical morphological and immunophenotypic features associated with t(8;21) acute myeloid leukemia.
Peer Review reports
Philadelphia (Ph)-negative myeloproliferative neoplasms (MPNs) are clonal hematopoietic disorders characterized by the proliferation of mature blood cells. Among them, polycythemia vera and essential thrombocythemia (ET) are the most frequent entities associated with chronic and indolent clinical course. Activating mutations of the JAK/STAT signal pathway, primarily JAK2 , MPL , and CALR , are present in the great majority of patients, exerting as primary pathogenesis leading to the proliferation of mature blood cells [ 1 ].
Blast transformation is a rare but well-recognized event in Ph-negative MPNs, associated with a grim prognosis. Secondary acute myeloid leukemias (AMLs) evolving from MPNs are characterized by a unique set of cytogenetic and molecular features distinct from de novo AML [ 2 , 3 , 4 , 5 ]. Somatic mutations in TP53 , ASXL1 , IDH1 / 2 , TET2 , and RUNX1 genes were reported to occur frequently in post-MPN AML, comparing with those in de novo disease [ 3 , 4 , 5 ]. On the contrary, structural alterations of chromosomes such as t(8;21)(q22;q22.1), inv(16)(p13.1q22)/t(16;16)(p13.1;q22), and t(15;17)(q22;q11-12), known as the most common cytogenetic aberrations in de novo AML, are quite rare in post-MPN AML [ 6 , 7 , 8 , 9 , 10 , 11 , 12 , 13 , 14 , 15 , 16 ]. Here we report a case of t(8;21) AML evolved from JAK2 -mutated ET.
A 72-year-old Japanese female patient was referred to the hospital because of thrombocytosis. She had no significant medical history. Peripheral blood showed hemoglobin 132 g/L, platelets 1046 × 10 9 /L, and white blood cells 8.4 × 10 9 /L. Bone marrow was hypercellular with increase of megakaryocytes (Fig. 1 ) and without increase of blasts (0.4%). Chromosomal analysis of bone marrow cells presented normal karyotype, and genetic tests revealed the presence of JAK2 V617F mutation. She was diagnosed with ET. Thrombocytosis had been well controlled by the administration of hydroxyurea; 2 years after initial diagnosis, her routine blood examination showed leukocytosis (white blood cells 14.0 × 10 9 /L with 82% of blasts), anemia (hemoglobin 91 g/L), and thrombocytopenia (platelets 24 × 10 9 /L). Bone marrow was hypercellular and infiltrated with 80% blasts, some of which possessed Auer rods (Fig. 2 A). These blasts were positive for myeloperoxidase staining (Fig. 2 B) and presented positivity for CD13, CD19, CD34, and CD56 on flow cytometry. G-banded chromosome analysis of bone marrow cells showed 46,XX,t(8;21)(q22;q22.1)[4]/45,idem,-X[16]. RUNX1::RUNX1T1 chimeric transcript was confirmed by reverse transcriptase polymerase chain reaction analysis. BCR::ABL1 fusion was not detected. She was diagnosed with AML with t(8;21)(q22;q22.1); RUNX1::RUNX1T1 .

Histopathology of bone marrow at the time of diagnosis with ET (hematoxylin and eosin, original magnification ×100), showing hypercellular marrow with increase of large to giant megakaryocytes. Megakaryocytes presented increased cytoplasm and hyperlobulated nuclei

A Morphologic features of blasts in bone marrow at the leukemic transformation (May–Giemsa staining, original magnification ×1000). Blasts contained fine azurophilic granules in abundant basophilic cytoplasm; arrow shows a blast with a single and sharp Auer rod. B Blasts were positive for myeloperoxidase staining
Allogeneic hematopoietic transplantation therapy was discussed, but she was considered to be transplant-ineligible because of higher age. Single usage of JAK inhibitor (ruxolitinib) was also clinically impractical as its efficacy in this setting was not established. As hypomethylating agent therapy was increasingly recognized as an alternative to induction chemotherapy for post-MPN AML [ 5 , 17 ], she was treated with combination chemotherapy by venetoclax and azacytidine. After the first cycle of the therapy, blasts disappeared from peripheral blood and decreased to 1.4% in bone marrow. During the course of the second cycle, she developed sepsis and pneumonia accompanying severe cardiac and respiratory failure. After the recovery from infections, she chose to be treated with best supportive care and the chemotherapy was discontinued.
For clonality analysis, RNA was prepared from CD34-sorted circulating leukemic blasts before chemotherapy and peripheral leukocytes after chemotherapy (65 days after the start of the therapy), consisting of 97% neutrophils, and subjected to reverse transcriptase polymerase chain reaction and nucleotide sequence analysis. As shown in Fig. 3 , RUNX1::RUNX1T1 chimeric transcript disappeared after chemotherapy, whereas heterozygous mutation of JAK2 V617F was present in both samples, indicating that the leukemic blasts were derived from JAK2 -mutated ET clone. Mutation of KIT or FLT-3 was not detected in leukemic blasts.

A RT-PCR detecting RUNX1::RUNX1T1 chimeric transcript. The sequences of primers were as follows; RUNX1 (forward), 5′- CTACCGCAGCCATGAAGAACC -3′; RUNX1T1 (reverse), 5′- AGAGGAAGGCCCATTGCTGAA -3′. Lane 1; leukemic blasts before chemotherapy, lane 2; peripheral leukocytes after chemotherapy. B Sequence analysis showing JAK2 V617F mutation. cDNA was amplified using primers, 5′-ATTTTTAAAGGCGTACGAAGAGAAGTAG-3′ (forward) and 5′-ATAAGCAGAATATTTTTGGCACATACAT-3′ (reverse). PCR product containing the reported mutation point was directly sequenced. Arrows indicate G > T substitution. 1; leukemic blasts before chemotherapy, 2; peripheral leukocytes after chemotherapy
Discussion and conclusion
Recent studies revealed that secondary AMLs evolving from Ph-negative MPNs present unique cytogenetic and molecular features distinct from de novo AML, including the higher frequency of somatic mutations in TP53 , ASXL1 , IDH1 / 2 , TET2, EZH2 , and RUNX1 genes and the lower frequency of those in FLT3 , MPM1 , CEBPA , and WT1 [ 3 , 4 ]. Chromosomal alterations related to poor-risk outcomes in MPNs include complex karyotypes, inv(3)(q21.3q26.2)/t(3;3)(q21.3;q26.2), i(17)(q10), -7/7q-, 12p-/12p11.2, or 11q23 rearrangements [ 5 ].
On the contrary, balanced chromosomal translocations frequently observed in de novo AML with good-risk complex are quite rare in post-MPN AML; 11 cases of acute promyelocytic leukemia with t(15;17)(q22;q11-12); PML::RARA evolved from MPNs were reported so far [ 6 , 7 , 8 , 9 , 10 , 11 , 12 , 13 , 14 , 15 ]. However, chromosomal rearrangements involving core binding factor genes, t(8;21) (q22;q22.1); RUNX1::RUNX1T1 or inv(16)(p13.1q22)/t(16;16)(p13.1;q22); CBFB::MYH11 were not reported, except for one case with ET-derived AML presenting t(8;21), in whom other gene mutations was not investigated [ 16 ]. Thus, to our best knowledge, the present case is the first one with JAK2 mutation preceding the acquisition of t(8;21).
Coexistence of JAK2 V617F mutation and t(8;21) chromosomal translocation is a rare but recurrent combination of genetic aberration in AML. JAK2 mutation has been reported to occur in a small number of de novo AML, with a relatively high incidence in subtype with t(8;21) associated with unfavorable clinical outcomes [ 18 , 19 , 20 ]. In these cases, JAK2 mutation was recognized as an additional genetic event after the formation of. RUNX1::RUNX1T1 chimeric gene. In our case, the opposite order of events, JAK2 mutation preceding the acquisition of t(8;21) was postulated, indicating that the occurrence of t(8;21) can be a late event in the progression of MPN, similarly as in a few cases with chronic myeloid leukemia with occurrent t(8;21) translocation and Ph chromosome [ 21 , 22 , 23 , 24 , 25 , 26 ].
Although high frequency of erythroblastic and megakaryoblastic phenotype was reported in post-MPN AML [ 27 ], our case presented typical morphological and immunophenotypic features associated with t(8;21) AML, such as the emergence of Auer rod and the expression of CD19, CD34, and CD56 [ 28 ]. The findings indicated that late-appearing cytogenetic abnormality would also define the phenotype of secondary AML from MPN.
Hydroxyurea, the most common cytoreductive agent employed in the management of MPN, interferes with DNA synthesis and may have mutagenic and leukemogenic potential. However, the recent study indicated that the use of hydroxyurea does not increase the risk of secondary malignancies, including AML and myelodysplastic syndrome [ 29 ]. Thus, it remains undetermined whether hydroxyurea promoted the formation of RUNX1::RUNX1T1 chimeric gene leading to leukemic transformation or not in this case.
Availability of data and materials
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
Myeloproliferative neoplasm
Acute myeloid leukemia
- Essential thrombocythemia
Philadelphia
Tefferi A, Pardanani A. Myeloproliferative neoplasms: a contemporary review. JAMA Oncol. 2015;1:97–105.
Article PubMed Google Scholar
Cerquozzi S, Tefferi A. Blast transformation and fibrotic progression in polycythemia vera and essential thrombocythemia: a literature review of incidence and risk factors. Blood Cancer J. 2015;5: e366. https://doi.org/10.1038/bcj.2015.95 .
Article CAS PubMed PubMed Central Google Scholar
Dunbar AJ, Rampal RK, Levine R. Leukemia secondary to myeloproliferative neoplasms. Blood. 2020;136:61–70.
Article PubMed PubMed Central Google Scholar
Luque Paz D, Jouanneau-Courville R, Riou J, et al . Leukemic evolution of polycythemia vera and essential thrombocythemia: genomic profiles predict time of transformation. Blood Adv. 2020;4:4887–97.
Tefferi A, Alkhateeb H, Gangat M. Blast phase myeloproliferative neoplasm: contemporary review and 2024 treatment algorithm. Blood Cancer J. 2023;13:108. https://doi.org/10.1038/s41408-023-00878-8 .
Batlle M, Fernández-Avilés F, Ribera JM, et al . Acute promyelocytic leukemia in a patient with idiopatic myelofibrosis. Leukemia. 1999;13:492–4.
Article CAS PubMed Google Scholar
Mollee PN, Taylor KM, Williams B, et al . Long-term molecular remission in promyelocytic transformation of myeloproliferative disease. Leukemia. 1999;13:648–50.
Kajiguchi T, Simokawa T, Saito M, et al . Transformation of polycythemia vera to acute promyelocytic leukemia. Int J Hematol. 2000;72:520–1.
CAS PubMed Google Scholar
Sato N, Furukawa T, Masuko M, et al . Acute promyelocytic leukemia developing in untreated essential thrombocythemia. Am J Hematol. 2002;71:114–6.
Braun TP, Maxson JE, Agarwal A, et al . Acute promyelocytic leukemia with JAK2 V617F and severe differentiation syndrome. Leuk Res Rep. 2015;4:8–11.
PubMed Google Scholar
Mamorska-Dyga A, Wu J, Khattar P, et al . Acute promyelocytic leukemia co-existing with JAK2 V617F positive myeloproliferative neoplasm: a case report. Stem Cell Investig. 2016;3:8.
Morsia E, Goteri G, Torre E, et al . Acute promyelocyte leukemia arose from CALR 1 mutated post essential thrombocythemia-myelofibrosis with splanchnic vein thrombosis: a case report. Leuk Res Rep. 2021;15: 100243.
CAS PubMed PubMed Central Google Scholar
Nadiminti K, Silverman M, Bhagavathi S, et al . t(15;17) associated with primary myelofibrosis: a case report of an unusual clinical presentation and diagnostic dilemma. Onco Targets Ther. 2019;12:5449–55.
Li W-W, Sui X-F, Fun S, et al . Transformation from polycythemia vera to acute promyelocytic leukemia: case report and literature review. Medicine. 2022;101: e30064. https://doi.org/10.1097/MD.0000000000030064 .
Zhang R, Liu R, Song H, et al . Clonal evolution analysis of a rare acute promyelocytic leukemia patient transforming from essential thrombocythemia. Ann Hematol. 2023;102:981–4.
Knottenbelt E, Hallett J, Jacobs P. 8;21 translocation in myelodysplasia secondary to essential thrombocythemia. Am J Hematol. 1989;30:233–5.
Gangat N, Guglielmelli P, Szuber N, et al . Venetoclax with azacytidine or decitabine in blast-phase myeloproliferative neoplasm: a multicenter series of 32 consecutive case. Am J Hematol. 2021;96:781–9.
Krauth M-T, Eder C, Alpermann T, et al . High number of additional genetic lesions in acute myeloid leukemia with t(8;21)/ RUNX1-RUNX1T1 : frequency and impact on clinical outcome. Leukemia. 2014;28:1449–58.
Iwanaga E, Nanri T, Matsuno N, Kawakita T, Mitsuya H, Asou N. A JAK2 -V617F activating mutation in addition to KIT and FLT3 mutations is associated with clinical outcome in patients with t(8;21)(q22;q22) acute myeloid leukemia. Haematologica. 2009;94:433–5.
Christen F, Hoyer K, Yoshida K, et al . Genomic landscape and clonal evolution of acute myeloid leukemia with t(8;21): an international study on 331 patients. Blood. 2019;133:1140–51.
Ferro MT, Steegman JL, Escribano L, et al . Ph-positive chronic myeloid leukemia with t(8;21)(q22;q22) in blastic crisis. Cancer Genet Cytogenet. 1992;58:96–9.
Kojima K, Yasukawa M, Ishimaru F, et al . Additional translocation (8;21)(q22;q22) in a patient with Philadelphia-positive chronic myelogenous leukaemia in the blastic phase. Br J Haematol. 1999;106:720–2.
Zhang Y, Liu Y, Liu X, et al . Co-existence of t(9;22) and t(8;21) in primary blast phase of chronic myelogenous leukemia: clinical experience and literature review. Int J Clin Exp Pathol. 2019;12:1811–5.
Ma C-C, Chai Y, Chen HL, et al . Clonal evolution of AML1-ETO coexisting with BCR-ABL and additional chromosome abnormalities in a blastic transformation of chronic myeloid leukemia. J Int Med Res. 2020. https://doi.org/10.1177/0300060520919237 .
Gong J-Y, Zhang Z-H, Zhang W, et al . Coexistence of recurrent chromosomal abnormalities and the Philadelphia chromosome in acute and chronic myeloid leukemias: report of five cases and review of literature. Mol Cytogenet. 2020;13:34–42.
Morita K, Jabbour E, Ravandi F, et al . Clinical outcomes of patients with concurrent core binding factor rearrangement and Philadelphia chromosome. Clin Lymphoma Myeloma Leuk. 2021;21:338–44.
Abdulkarim K, Girodon F, Johansson P, et al . AML transformation in 56 patients with Ph- MPD in two well defined populations. Eur J Haematol. 2009;82:106–11.
Arber DA, Porwit A, Brunning RD, et al . Acute myeloid leukaemia with recurrent genetic abnormalities. In: Swedlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, editors., et al ., World Health organization classification of tumours of haematopoietic and lymphoid tissues. 4th ed. Lyon: Lyon IARC Press; 2017. p. 130–49.
Google Scholar
Wang R, Shallis RM, Stempel JM, et al . Second malignancies among older patients with classical myeloproliferative neoplasms treated with hydroxyurea. Blood Adv. 2023;7:734–43.
Download references
Acknowledgements
We thank Dr. Yuichi Sugahara, Higashimatsuyama Municipal Hospital, for lending BM biopsy specimen at the diagnosis with ET.
There were no funding sources to support this study.
Author information
Authors and affiliations.
Department of Hematology, Saitama Medical University Hospital, 38 Morohongo, Iruma-gun, Moroyama, Saitama, 350-0495, Japan
Chie Asou, Tomoyuki Sakamoto, Kodai Suzuki, Itoko Okuda, Atsushi Osaki, Ryohei Abe, Yoshihiro Ito, Emi Kakegawa, Yoshitaka Miyakawa, Yasuhito Terui & Yuichi Nakamura
You can also search for this author in PubMed Google Scholar
Contributions
CA wrote the manuscript and took care of the patient. TS, KS, IO, AO, RA, and YI edited the manuscript and took care of the patient. EK performed molecular analysis. YM and YT reviewed and edited the manuscript. YN wrote and edited the manuscript. All authors approved the final manuscript and consented to publish the manuscript.
Corresponding author
Correspondence to Yuichi Nakamura .
Ethics declarations
Ethical approval and consent to participate.
The study was performed with prior approval of the Institutional Review Board and the Ethics Committee of the Saitama Medical University Hospital (the committee’s reference number: 12-089). Written informed consent for the study was obtained from the patient.
Consent for publication
Written informed consent was obtained from the patient’s legal guardian for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s note.
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/ .
Reprints and permissions
About this article
Cite this article.
Asou, C., Sakamoto, T., Suzuki, K. et al. Transformation into acute myeloid leukemia with t(8;21)(q22;q22.1); RUNX1::RUNX1T1 from JAK2 -mutated essential thrombocythemia: a case report. J Med Case Reports 18 , 372 (2024). https://doi.org/10.1186/s13256-024-04691-0
Download citation
Received : 09 December 2023
Accepted : 22 June 2024
Published : 18 August 2024
DOI : https://doi.org/10.1186/s13256-024-04691-0
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
- Myeloproliferative neoplasms
- Secondary acute myeloid leukemia
- t(8;21)(q22;q22.1)
- RUNX1::RUNX1T1
Journal of Medical Case Reports
ISSN: 1752-1947
- Submission enquiries: Access here and click Contact Us
- General enquiries: [email protected]

- Get new issue alerts Get alerts
Secondary Logo
Journal logo.
Colleague's E-mail is Invalid
Your message has been successfully sent to your colleague.
Save my selection
Acute myeloid leukemia in an 86-year-old man with AML1/ETO treated with Homoharringtonine and Arsenic Trioxide
A case report.
Editor(s): NA.,
Department of Hematology, Union Hospital, Fujian Medical University, Fujian Institute of Hematology, Fujian Provincial Key Laboratory of Hematology, Fuzhou, China.
∗Correspondence: Yong Wu, Department of Hematology, Fujian Medical University Union Hospital, Fujian Institute of Hematology, 29 Xinquan Road, Fuzhou 350001, China (e-mail: [email protected] ).
Abbreviations: AML = acute myeloid leukemia, AML-M2 = AML with maturation, As 2 O 3 = Arsenic Trioxide, Hb = hemoglobin, HHT = Homoharringtonine, PLT = platelet, WBC = white blood cell.
Current publication is supported by grants from the Construction project of Fujian Medical Center of Hematology (Min201704), National and Fujian Provincial Key Clinical Specialty Discipline Construction Program, China (2011-1006, 2012-149).
The authors have no conflicts of interest to disclose.
This is an open access article distributed under the terms of the Creative Commons Attribution-Non Commercial License 4.0 (CCBY-NC), where it is permissible to download, share, remix, transform, and buildup the work provided it is properly cited. The work cannot be used commercially without permission from the journal. http://creativecommons.org/licenses/by-nc/4.0
Rationale:
Acute myeloid leukemia (AML) is a malignantly clonal and highly heterogeneous disease. Although the treatment of AML has brought promising outcomes for younger patients, prognosis of the elderly remains dismal. Innovative regimens are increasingly necessary to be investigated.
Patient concerns:
We present an 86-year-old AML patient with fever, cough, and sputum production.
Diagnoses:
A diagnosis of AML with maturation (AML-M2) and AML1/ETO was made.
Interventions:
The patient was treated with a regimen of Homoharringtonine coupled with arsenic trioxide.
Outcomes:
The AML-M2 patient with AML1/ETO achieved incomplete remission, but showed few toxic side effects and improved survival. Besides, we analyzed the dynamic counts of complete blood cells during the treatment. The count of white blood cell had a positive correlation with the percentage of blast cells ( r = 0.65), both of which had a negative correlation with the percentage of segmented neutrophils ( r = –0.63, –0.89).
Lessons:
Homoharringtonine and arsenic trioxide may induce both the apoptosis and differentiation of leukemic cells in AML-M2 with AML1/ETO.
1 Introduction
Acute myeloid leukemia (AML) is a malignantly clonal disorder characterized by blockage of differentiation in the myeloid lineage and an accumulation of its immature progenitors in bone marrow, leading to hematopoietic failure. [1] In China, it was predicted that there were about 75,300 newly diagnosed leukemia cases in 2015; meanwhile, it was estimated that about 53,400 Chinese died of leukemia in 2015. [2] Age has been recommended as one of the poorest prognostic indicators for overall survival over the past decades. Although the changing treatment schedules and transplantation have shown benefits in AML of younger patients, response to treatment and survival in older ones remains dismal. [3] Here, we reported a successful case of 86-year-old man with AML treated with traditional Chinese medicines (TCM), Homoharringtonine and Arsenic, showing few toxic side effects and improved survival.
This study was approved by Ethical Committee of Union Hospital Affiliated to Fujian Medical University (2018YF037-02), and written informed consent was obtained from the patient's family for publication of this case report and accompanying images.
3 Case presentation
An 86-year-old man with fever, cough and sputum production for 7 days, was admitted to our hospital in November 2016. The medical history revealed the patient diagnosed with malignant lymphoma by the biopsy of cervical lymph node 4 years ago, had received 6 courses of standard chemotherapy (CHOP regimen), and had 5 years history of diabetes. Apart from the signs of anemia in the aged man, peripheral blood counts revealed white blood cells (WBC) 40.05 × 10 9 /L, segmented neutrophils 2%, hemoglobin (Hb) 76.0 g/L, platelet (PLT) 74.0 × 10 9 /L, and blast cells accounted for 90% of nucleated cells. Bone marrow was examined in an effort to establish the diagnosis, showing a marked hypercellularity with 68% myeloblasts, the occurrence of Auer rods, and 100% positive myeloperoxidase staining. AML1-ETO fusion gene was also detected. Consequently, the elderly patient was diagnosed with AML-M2 based on French–American–British classification.
He was treated with Homoharringtonine 2 mg/d and arsenic trioxide (As 2 O 3 ) 10 mg/d after the initial diagnosis. But Homoharringtonine and As 2 O 3 were replaced by supportive therapy due to overt myelosuppression 4 days later. Peripheral blood examination revealed WBC 1.71 × 10 9 /L (myeloblasts decreased to 25% and segmented neutrophils increased to 51% of all nucleated cells), Hb 44.0 g/L, and PLT 13.0 × 10 9 /L. Surprisingly, no myeloblast was detected and segmented neutrophils were 34% at day 9 after the chemotherapy. Whereas the follow-up count of WBC increased to 73.43 × 10 9 /L and myeloblasts increased to 97% at day 47 after his first chemotherapy. The initial regimen of Homoharringtonine and As 2 O 3 were reused. The count of WBC returned to normal 3 days later and the chemotherapy was then discontinued. In order to reduce the degree of myelosuppression, we chose the regimen of As 2 O 3 between 5 mg × 7 day and 10 mg × 7 day, alternately. Meanwhile, the regimen of Homoharringtonine between 0.5 mg × 7 day and 1 mg × 7 day was adopted, alternately. No myeloblast was detected in the peripheral blood cell smear with myelocytes 23%, metamyelocytes 22%, and segmented neutrophils 51% after 2 courses of the regimen above.
Analyzing the correlations among complete blood cell counts with Spearman test [4] in our case, we found some features as follows: The patient displayed an abnormally elevated count of WBC, and aberrantly decreased counts of PLT and Hb at his first visit, which was consistent with pathological feature of AML. Besides, the count of WBC had a positive correlation with the percentage of blast cells ( r = 0.65), but a negative correlation with the percentage of segmented neutrophils ( r = –0.63). The percentage of blast cells had a negative correlation with the percentage of segmented neutrophils ( r = –0.89). It may be explained by the differentiation from blast cells to segmented neutrophils after chemotherapy. However, the counts of PLT and Hb had no correlation with the other parameters above ( Fig. 1 ).

4 Discussion
Usually, AML patients have no evident causes. Exposure to chemotherapy is 1 risk factor associated with increased incidence with age. In our case, the patient with lymphoma had received chemotherapy for 6 cycles before the diagnosis of AML-M2, the cause of which may be the chemotherapy. In addition, AML1-ETO fusion gene was found in the case diagnosed with AML-M2. Whether the occurrence of AML1-ETO gene is before lymphoma or not, is not known. AML1-ETO gene is the product of t(8;21)(q22;q22) translocation in AML patients. AML1-ETO keeps the function of DNA binding sites in AML1 and the ability to recruit relevant cofactors through ETO, promoting granulopoiesis with inhibition of erythropoiesis in bone marrow. [5]
Older AML patients (age >60 years) have always been one of the most challenging group to treat. These patients have different tolerance of toxicity, and treatments are hardly curative. Treatment-related mortality of elderly patients with intensive treatment is more common (10%–40%) than that of younger patients (<10%). [1] Innovative chemotherapy regimens are thus necessary to be investigated. A randomized controlled, phase 3 trial study in 609 AML patients (14–59 years old) from China reported that the Homoharringtonine based HAA regimen (Homoharringtonine, aclarubicin, and cytarabine) had a higher complete remission (CR) rate and survival advantage than the daunorubicin and cytarabine regimen. [6] More recently, a retrospective research of 140 patients (16–60 years old) with t(8;21)AML revealed that the HAA regimen provided good molecular response and achieved much higher CR rate after 1 cycle of induction treatment, compared to other regimens reported in t(8;21)AML. [7] Thus, the Homoharringtonine based regimen may be a better choice in AML, especially in t (8; 21) AML. Arsenic, with a 500-year history in TCM, was successfully used to treat acute promyelocyte leukemia (APL) in TCM principle of counteracting one toxin with another. [8,9] In 2000, Chinese researchers reported that the CR rate and the 5-year survival rate of 136 APL patients treated with As 2 O 3 were 87.9% and 92.0%, respectively. [10]
On the basis of the encouraging results above, we selected Homoharringtonine coupled with As 2 O 3 to treat the 86-year-old case diagnosed as AML-M2 with AML1-ETO fusion gene, and a good response was achieved after the 2-cycle chemotherapy. We observed leukocytes decreased rapidly and blast cells differentiated to segmented neutrophils after chemotherapy. The antileukemic effects of Homoharringtonine mainly depended on inhibiting protein synthesis to inhibit proliferation, induce differentiation, and promote apoptosis of leukemic cells, leukemia stem cells included too. [11–15] Moreover, Chen et al [9] found that As 2 O 3 mediated a dual effect on APL cells in a dose-dependent manner in vitro and vivo studies. A higher concentration of As 2 O 3 (0.5–2.0 pmol/L) led to apoptosis which was associated with mitochondrial pathway and the degradation of PML-RARα oncoprotein, while a lower concentration of As 2 O 3 (0.1–0.5 pmol/L) induced partial differentiation related to granulocytic pathway to some extent. We observed the leukocytes reduction and blast cells differentiation after chemotherapy. Additionally, Chinese investigators reported that all-trans retinoic acid could induce differentiation in t(8;21) AML leukemic cells. [16] But the underlying mechanisms of Homoharringtonine and As 2 O 3 are still needed to be elucidated in AML1-ETO positive cell lines.
5 Conclusion
To conclude, the regimen of Homoharringtonine coupled with As 2 O 3 may bring substantial effects on elderly AML-M2 patients, which must rely on randomized controlled trials on many more patients to confirm. Besides, more experiments on AML1-ETO – expressing cell lines should be carried out to understand the potential mechanisms.
Author contributions
Conceptualization: Zhipeng He.
Data curation: Meiling Chen, Lili Chen, Bixin Wang.
Investigation: Yiping Huang, Huixian Wang, Mengting Yang, Jiaying Chen.
Writing – original draft: Zhipeng He.
Writing – review and editing: Zhipeng He, Xueting Xiao, Yanhong Lu, Yong Wu.
- Cited Here |
- Google Scholar
acute myeloid leukemia; AML1/ETO; Arsenic Trioxide; Homoharringtonine
- + Favorites
- View in Gallery
- Current Issue
- Supplements
The Pure Erythroleukemia: A Case Report and Literature Review
Naeem latif, md, elaine salazar, md, rubina khan, md, bruce villas, md, fauzia rana, md, the pure erythroleukemia: a case report and literature review.
Naeem Latif, MD, Elaine Salazar, MD, Rubina Khan, MD, Bruce Villas, MD, Fauzia Rana, MD
Department of Hematology/Oncology and Pathology University of Florida College of Medicine Jacksonville, Florida
Introduction
Pure acute erythroleukemia is a rare form of acute myeloid leukemia with predominant erythroid lineage proliferation. It is a heterogeneous entity amongst acute myeloid leukemia (AML) that can occur at any age, including childhood, and comprises less than 5% of AML. Di Guglielmo reported the original case of acute erythroleukemia in 1917; he described it as a syndrome composed of immature erythroid and myeloid elements characterized by a pure normoblastic proliferation.1,2
Classification
In 1985, the French-American-British (FAB) Cooperative group revised its criteria by requiring at least 30% of the non-erythroid elements to be type I or II blasts, and defined AML-M6 as “a proliferation of more than 50% erythroblast and more than 30% of myeloblasts within non-erythroid cells.”3 According to new World Health Organization (WHO) classification, 2 subtypes are recognized based of the presence or absence of a significant myeloid (granulocytic) component. The first subtype, pure erythroid leukemia (FAB subtype B), represents a neoplastic proliferation of immature cells (undifferentiated or proerythroblastic in appearance) committed exclusively to the erythroid lineage (>80% of bone marrow cells) with no evidence of a significant myeloblastic component. The second subtype, erythroleukemia (erythroid/myeloid FAB subtype A), is defined by the presence in the bone marrow of more than 50% erythroid precursors in the entire nucleated cell population and more than 20% myeloblasts in the nonerythroid cell population (the myeloblasts are calculated as a percent of the non-erythroid cells).4
Case Report
A 57-year-old African American woman presented to the emergency room with shortness of breath and chest pain. The patient complained of fatigue, generalized body aches, and pain during the previous weeks. She denied fever, chills, weight loss, or night sweats. The patient had no significant medical history and was not taking any medications. She had no family history of malignancy; she smoked one pack daily for many years and had a history of heavy alcohol use in the distant past, but denied any drug abuse. The review of systems was negative for fever, chills, weight loss, night sweats, ecchymosis, or bleeding. Physical examination revealed a pale-looking woman without any obvious distress. She was afebrile with normal vitals. There were no petechiae, ecchymosis, or gum bleeding. There was no palpable lymphadenopathy or hepatosplenomegaly, and the rest of the physical examination was normal.
Pathologic Findings and Hospital Course
The patient’s initial complete blood count profile showed pancytopenia, with a white blood cell count of 3.3 × 103/μL, a hemoglobin level of 4.4 g/dL, and a platelet count of 29.0 × 103/μL. Numerous nucleated red blood cells, occasional erythroblasts, and a rare circulating megakaryoblast (Figure 1A) were identified on peripheral smear examination. Examination of the initial bone marrow aspirate and biopsy revealed a markedly hypercellular bone marrow essentially replaced by erythroid precursors, representing approximately 80–90% of the marrow cells (Figure 1B). The erythroid precursors displayed dysplastic morphology, including megaloblastic features, multinucleation, and vacuolization, and they were strongly periodic acid-Schiff (PAS)–positive (Figure 1C). Occasional myeloid and megakaryocytic elements were encountered with myeloblasts representing less than 5% of marrow cells. These findings are consistent with pure erythroid leukemia. Flow cytometric analysis of the bone marrow aspirate also revealed numerous immature cells, which were positive for CD 36 and glycophorin A, consistent with erythroblasts/erythroid precursors. Only 3% of the total cells analyzed were CD34/CD13-positive myeloblasts. These flow cytometric and immunophenotypic findings also supported the diagnosis of pure erythroid leukemia (acute erythroid leukemia), FAB subtype M6b. Significant cytogenetic abnormalities were found on chromosomal analysis, including a complex karyotype (45xx, der(5)t(5;17)(q13;q11.2),r(7)(p14q36),-16,-17,add(19) (q13.4)+mar(14)/46xx(6) without the typical deletions of chromosomes 5 and 7 that are often seen in myelodysplastic syndrome and associated with poor prognosis. The patient was treated with a standard induction (7+3) chemotherapy regimen consisting of daunorubicin 45 mg/m2 daily for 3 days and cytarabine 100 mg/m2 daily continuous infusion for 7 days. She tolerated treatment well and required blood and platelet transfusions. On post-induction day 14, the bone marrow biopsy showed residual disease with 10% erythroblasts, treatment effects, and abnormal myeloid precursors. Cytogenetics showed persistent multiple abnormalities including 42-44,XX, -3,der(5;17)(q13;q11.2),r(7)(p14q36),-15,-17,+mar(cp3)/46,XX.17
The patient received 3 more cycles of consolidative high-dose cytarabine chemotherapy without complete cytogenetic or molecular remission. The treatment regimen was switched to mitoxantrone and etoposide, but the patient’s disease transformed into AML. Four months after the initial bone marrow examination and treatment, the patient converted to a true AML. Flow cytometric analysis of the bone marrow aspirate revealed abnormal vacuolated erythroblasts, which were positive for CD 36 and glycophorin A, consistent with residual erythroleukemia. In addition, there was now a significant myeloblast population (30%) of total cells analyzed, which were positive for CD 13 and CD 34, which had increased from the first presentation of 3%.
Histologically, the bone marrow was hypercellular and virtually replaced by erythroid and myeloid precursors representing approximately 70–80% of marrow cells. The erythroid precursors continued to display dysplastic morphology. Numerous smaller vacuolated blasts consistent with myeloblasts with therapy effect and occasional megakaryocytic elements were also seen. Further supporting the transformation of pure erythroleukemia to AML was a new cytogenetic finding: the acquisition of trisomy of chromosome 8 (+8), the most frequent trisomy in malignant myeloid disorders. Trisomy 8 is also associated with the development of myeloid blast phase chronic myelogenous leukemia (CML).
The patient could not achieve complete remission and opted for supportive care for her refractory disease;she expired 5 months after diagnosis.
Clinical Presentation
The incidence of acute erythroid leukemia is rare and ranges from 3% to 8% with an approximate 50% chance of patients developing either de novo or secondary erythroleukemia.5-8 There is a male preponderance, and age distribution appears to be bimodal, with a smaller peak below 20 years and a more definitive and broader peak in the seventh decade of life.9 In almost 50% of cases, AML-M6 occurs secondary to chemotherapy with alkylating agents or occupational exposure to mutagenic agents (eg, benzene exposure). Our patient had a remote history of heavy alcohol abuse, consistent with some reports of alcoholism and erythroid leukemia. AML-M6 may also develop as a blast crisis of myeloproliferative disease or a final evolution of myelodysplastic syndrome.
The most common complaint in patients with eythroid leukemia is severe anemia, which is present in most patients and is often severe (mean hemoglobin, 7.5 g/dL). One-third of the patients were reported to have hemorrhage. The presence of hepatosplenomegaly varies between 20–40%.10 Fifty percent of the patients have no blasts on the peripheral blood smear. Neutrophil and platelet counts are mildly to markedly diminished. Peripheral blood findings may also include a high level of schistocytes, nucleated red cells, and pseudo Pelger-Huet neutrophils. However, these abnormalities are not specific for erythroleukemia and can present in other dyserythropoiesis etiologies such as myelodysplastic syndrome.
The bone marrow is usually hypercellular and shows major dysplasia in the red cells. Erythroid lineage is dysplastic, with megaloblastoid cells (asynchronous nucleocytoplasmic maturation), Howell-Jolly bodies, and lack of hemoglobinization and multinucleation. Multilineage dysplasia is present in most reports.10 Megakaryocytes are often dysplastic, with abnormalities of segmentation of the nucleus or abnormalities of size (micromegakaryoblasts); rarely, megakaryoblasts may circulate in the peripheral blood. Dysgranulopoiesis is described in 50% of patients.11 This heterogeneity of trilineage dysplasia can be explained by the fact that erythroleukemia includes both primary and secondary diseases.
Morphology and Cytochemistry
Pure Erythroid Leukemia (FAB Subtype B)
The undifferentiated form of pure erythroid leukemia is usually characterized by the presence of medium to large erythroblasts usually with round nucleoli (proerythroblast); the cytoplasm is deeply basophilic, often agranular, and frequently contains poorly demarcated vacuoles that are often PAS-positive. Occasionally, the blasts are smaller and resemble the lymphoblasts of ALL. The cells are negative for myeloperoxidase; they show reactivity with alphanaphthyl acetate esterase, acidic phosphatase, and PAS; the latter usually in a block-like staining pattern. In the bone marrow biopsies of pure acute erythroid leukemia, the cells appear undifferentiated.4 4
A large variety of cytochemical stains are available for characterization of erythroid lineage. The PAS reaction is always negative in normal erythroid differentiation. Aberrant PAS positivity is often observed in the pronormoblasts and basophilic normoblasts in AML-M6. The Prussian blue stain demonstrates increased iron stores in AML-M6, sometimes with ringed sideroblasts. Some authors reported occasional positivity of myeloperoxidase in erythroid cells of erythroleukemia.12
Flow Cytometry and Immunophenotyping
The erythroblasts in erythroleukemia generally lack myeloid-associated markers and are negative with antimyeloperoxidase stains. The best known markers for erythroleukemia have included glycophorin A and CD36. However, glycophorin A has been reported to be completely negative in some cases of AML-M6b, probably because it is a late erythroid marker. CD36, a nonspecific marker, detects erythroid precursors at earlier stages of differentiation and is also expressed by monocytes and megakaryocytes. An aberrantly low expression of CD71 may be present in erythroleukemia; a nonspecific activation marker can be present in other AML cases. The immunophenotype of the myeloid population usually corresponds to that of AML without differentiation or AML with minimal differentiation and is now called pure erythroid leukemia in the new WHO classification.4,13-15
The more differentiated form of pure erythroid leukemia can be detected by the expression of glycophorin A, the absence of myeloperoxidase, and other myeloid markers. The blasts are often negative for HLA-DR and CD34, but may be positive for CD117.
Cytogenetics
A large array of chromosomal abnormalities has been described in AML-M6b, but no consistent pattern has been specified. Clonal chromosomal abnormalities are found in 70–100% of patients. This heterogeneity is probably due to inclusions of secondary or therapy-related AML-M6 in some series. Loss of all or part of the long arm, -5/del(5q), -7/del(7q), and trisomy 8 were observed in approximately 65% of de novo and secondary AMLM6.
In only 1 series, the overall frequency of the abnormalities of chromosomes 5 and/or 7 observed in de novo AML-M6 patients is similar to that observed in patients with therapy-related AML and substantially higher than that noted in patients with other types of de novo AML.16 In every series, the incidence of patients with an unfavorable karyotype is higher than in other types of AML with inv 3, 11q23, and 17p abnormalities.17 However, the cases with -5/Del (5q), -7/Del (7q), and/or complex chromosomal abnormalities should be classified as AML with myelodysplastic-related changes if the other requirements for that category are satisfied. The karyotypes are often complex, with multiple abnormalities and subclones. Using fluorescence in situ hybridization, some cases of AML-M6 clearly demonstrated similar karyotypic abnormalities within both myeloid and erythroid cells. Atkinson and colleagues proposed that a characteristic of this disease may be the presence of an altered primitive cell with features of both erythroid and myeloid lineages.16 This theory may provide a possible explanation for why many cases of acute erythroid leukemia progress to AML, as was observed in our patient.
Differential Diagnosis
The erythroleukemia (erythroid/myeloid) should be distinguished from refractory anemia with excess blasts, AML with myelodysplasia-related changes, AML with increased erythroid precursors, and reactive erythroid hyperplasia following therapy or administration of erythropoietin. A bone marrow biopsy with differential count of all nucleated cells should be performed. If the overall percentage of blast cells is more than 20% and multilineage dysplasia is present in more than 50% of the cells in 2 or more lineages, a diagnosis of AML with myelodysplasiarelated changes should be made. When there are less than 20% total blasts and the erythroid precursors are more than 50% of all cells, the differential count of nonerythroid cells should be calculated. If the blasts are more than 20% of nonerythroid cells, the diagnosis is erythroleukemia (erythroid/myeloid); if they are less than 20%, the diagnosis is usually myelodysplastic syndrome.
The differential diagnosis of pure erythroleukemia includes megaloblastic anemia due to vitamin B12 deficiency. Pure erythroid leukemia without morphologic evidence of erythroid maturation may be difficult to distinguish from other types of AML, particularly megakaryoblastic leukemia, and also from acute lymphoblastic leukemia and lymphoma. Lack of expression of lymphoid markers will exclude the latter diagnosis.4
Treatment and Prognosis
The outcome is usually very poor for erythroleukemia. In de novo AML-M6b, treatment with intensive chemotherapy (anthracycline and cytarabine) gives a complete remission (CR) rate of approximately 50–60%.10 In other series, the CR rate was only 10–40%, especially in secondary AML-M617; if CR was obtained, it was brief. The median survival in AML-M6B is approximately 4–5 months, and survival is related to karyotypic abnormalities.10 Allogeneic bone marrow transplantation remains the best treatment option for patients with abnormalities of 5q or 7q because they carry a worse prognosis.18 Even for therapy-related erythroleukemia, transplantation seems to be favorable for long-term disease-free survival.19 The response to chemotherapy and the length of survival is dependent on many factors, the most important of which is cytogenetics and karyotype abnormality.16,20-22 The CR rate in patients with 5q or 7q abnormalities is approximately 20%, and median survival reaches 16 weeks, compared to 77 weeks for patients without these abnormalities.10 The difference in prognosis between de novo and secondary leukemia is related to karyotype abnormalities. According to Kowal-Vern and associates, the percentage of pronormoblasts appears to be an important factor for survival, with a mean survival of 34 months in AML-M6a, 3.5 months in AML-M6b, and 10.5 months for AML-M6c.12 Age is another important prognostic factor; older patients have a low CR and a short survival time. Expression of P-glycoprotein is clearly a poor prognostic factor for survival.11 Other prognostic factors include good performance status, normal organ function, de novo presentation, and lack of multidrug resistance (MDR) expression.23 The management of AML-M6b subtype for younger patients (eg, 55–65 years) with good performance status is an induction chemotherapy (7+3) regimen (7 days of continuous infusion of cytarabine [ara C] and 3 days of anthracycline); 2–6 cycles of consolidation with highdose cytarabine are given if CR occurs.
Cytarabine remains the most active agent in the management of AML, and various regimens are designed to be combined with this agent. The dose of induction chemotherapy with cytarabine, anthracycline (idarubicinor daunorubicin) or anthracenedione (mitoxantrone) varies, but similarities outweigh the differences. There are 2 options for consolidation therapy: the high-dose ara-C regimen consists of intravenous ara-C3 g/m2 every 12 hours on days 1, 3, and 5, and the 5 +2 regimen includes ara-C at 100 mg/m2/day continuous infusion on days 1–5 plus intravenous anthracycline at 45–90 mg/m2 on days 1 and 2. The new dosing recommendations for anthracycline as induction chemotherapy are up to 90 mg/m2. A bone marrow biopsy should be performed 14–21 days after induction chemotherapy to assess remission status. If bone marrow shows persistent blasts, re-induction with the 5+2 regimen is recommended. If the bone marrow is hypoplastic, the second course shouldbe delayed until the bone marrow is recovered enough to clearly distinguish the type of recovery (leukemic vs normal marrow). The recovering bone marrow usually shows immature cells, which helps to distinguish them from myeloblast immunophenotyping; flow-cytometry and cytogenetics are helpful to detect residual disease.
AML-M6 is a heterogeneous disease with poor response to standard chemotherapy that caries a poor prognosis. The new WHO classification subdivides acute erythroid leukemia into erythroleukemia (erythroid/myeloid) and pure erythroid leukemia. AML-M6 appears to be bilineage leukemia; our case initially presented as pure erythroleukemia with 80–90% erythroblasts and less than 5% myeloblasts, but after treatment with chemotherapy, it transformed into AML with an increase in myeloblasts up to 30%. Our patient transformed from pure erythroleukemia to AML after treatment probably due to the erythroid component of leukemia achieving remission and the myeloid component remaining refractory to therapy and becoming predominant. Erythroleukemia usually presents with pancytopenia and mostly carries complex cytogenetics, including abnormalities of chromosomes 5, 7, and 8. There are other poor-risk molecular abnormalities like the presence of P-glycoprotein, Flt3 mutation,24 Kit mutation,25 and high MRD. Flt3 and Kit mutations can be useful for therapeutic implication; Flt3 inhibitors are in phase I/II trials with some promising results. The new higher dose of anthracycline (90 mg/m2) should be used for induction chemotherapy if cardiac function is normal. Allogeneic bone marrow transplant should be considered upfront for appropriate candidates once remission is achieved in AML-M6, since the risk of relapse and mortality is very high with this disease.
1. Di Guglielmo G. Le Maltase Eritremiche Ed Eritroleucemiche. II Pensiero Scientifico. Haematologica. 1928;9:301-347.
2. Roggli VL, Saleem A. Erythroleukemia: a study of 15 cases and literature review.Cancer. 1982;49:101-108.
3. Bennett JM, Catovsky D, Daniel MT, et al. Proposed revised criteria for the classification of acute myeloid leukemia. A report of the French-American-British Cooperative Group. Ann Intern Med. 1985;103:620-625.
4. Swerdlow SH, Campo E, Harris NL, et al, eds. WHO Classification of Tumors of Hematopoietic and Lymphoid Tissues (IARC WHO Classification of Tumours). 4th ed. Lyon, France: WHO Press; 2008.
5. Goldberg SL, Noel P, Lumpp TR, Dewald G. The erythroid leukemias: a comparative study of erythroleukemia (FABM6) and Di Guglielmo disease. Am J Clin Oncol. 1998;21:42-47.
6. Wheatley K, Burnett AK, Goldstone AH, et al. A simple, robust, validated and highly predictive index for the determination of risk directed therapy in acute myeloid leukemia derived from the MRC AML10 trial. Br J Haematol. 1999;107: 69-79.
7. Davey FR, Abraham N, Brunetto VL, et al. Morphologic characteristics of erythroleukemia (acute myeloid leukemia; FAB-M6): a CALGB study. Am J Hematol. 1995;49:29-38. 6 Clinical Advances in Hematology & Oncology Volume 8, Issue 4 April 2010
8. Büchner T, Hiddemann W, Wörmann B, et al. Double induction strategy for acute myeloid leukemia: the effect of high-dose cytarabine with mitoxantrone instead of standard-dose cytarabine with daunorubicin and 6-thioguanine: a randomized trial by the German AML cooperative group. Blood. 1999;93:4116-4124.
9. Kowal-Vern A, Mazzella FM, Cotelingam JD, et al. Diagnosis and characterization of acute erythroleukemia subsets by determining the percentages of myeloblasts and proerythroblasts in 69 cases. Am J Hematol. 2000;65:5-13.
10. Olopade OI, Thangavelu M, Larson RA, et al. Clinical, morphologic and cytogenetic characteristics of 26 patients with acute erythroblastic leukemia. Blood. 1992;80:2873-2882.
11. Mazzella F, Alvares C, Kowal-Vern A, Schumacher H. The acute erythroleukemias. Clin Lab Med. 2000;20:119-137.
12. Kowal-Vern A, Cotelingam J, Schumacher HR. The prognostic significance of proerythroblasts in acute erythroleukemia. Am J Clin Pathol. 1992;98:34-40.
13. Southcott MJ, Tanner MJ, Anstee DJ. The expression of human blood group antigens during erythropoiesis in a cell culture system. Blood. 1999;93:4425-4435.
14. Garand R, Duchayne E, Blanchard D, et al. Minimally differentiated erythroleukemia (AML M6 ‘variant’): a rare subset of AML distinct from AML M6. Groupe Francais d Hematologie Cellulaire. Br J Haematol. 1995;90:868-875.
15. Jaffe ES, Harris NL, Stein H, Vardiman JW, eds. Pathology and Genetics: Tumours of Haematopoietic and Lymphoid Tissues (WHO Classification of Tumours). Lyon, France: IARC Press; 2001.
16. Atkinson J, Hrisinko MA, Weil SC. Erythroleukemia: a review of 15 cases meeting 1985 FAB criteria and survey of the literature. Blood Rev. 1992;6:204-214.
17. Park S, Picard F, Azgui Z, et al. Erythroleukemia: a comparison between the previous FAB approach and the WHO classification. Leuk Res. 2002;26:423-429.
18. Sutton L, Chastang C, Ribaud P, et al. Factors influencing outcome in de novo myelodysplastic syndromes treated by allogeneic bone marrow transplantation: a long term study of 71 patients. Societe Francaise de Greffe de Moelle. Blood. 1996;88:358-365.
19. Ballen KK, Gilliland DG, Guinan EC, et al. Bone marrow transplantation
for therapy-related myelodysplasia: comparison with primary myelodysplasia. Bone Marrow Transplant. 1997;20:737-743.
20. Nakamura H. Cytogenetic heterogeneity in erythroleukemia defined as M6 by the French-American-British Cooperative Group criteria. Leukemia. 1989;3: 305-309.
21. Cuneo A, Van Orshoven A, Michaux JL, et al. Morphologic, immunologic and cytogenetics studies in erythroleukemia: evidence for a multilineage involvement
and identification of two distinct cytogenetic-clinicopathological types. Br J Haematol. 1990;75:346-354.
22. Kottaridis PD, Gale RE, Frew ME, et al. The presence of FLT3 internal tandem duplication in patients with acute myeloid leukemia (AML) adds important prognostic information to cytogenetic risk group and response to the first cycle of chemotherapy: analysis of 854 patients from the United Kingdom MedicalResearch Council AML 10 and 12 trials. Blood. 2001;98:1752-1759.
23. Mazzella FM, Kowal-Vern A, Shirt A, Rector JT, Cotelingam JD, Schumacher HR. Effects of multidrug resistant gene expression in acute erythroleukemias. Mod Pathol. 2000;13:407-413.
24. Whitman SP, Ruppert AS, Radmacher MD, et al. FLT3 D835/I836 mutations are associated with poor disease-free survival and a distinct gene-expression signature among younger adults with de novo cytogenetically normal acute myeloid leukemia lacking FLT3 internal tandem duplications. Blood. 2008; 111:1552-1559.
25. Schnittger S, Kohl TM, Haferlach T, et al. KIT-D816 mutations in AML1-ETO-positive AML are associated with impaired event-free and overall survival. Blood. Mar 2006;107:1791-1799.
Erythroleukemia: Clinical Course and Management
Aisha Masood,1 Beata Holkova,2 and Asher Chanan-Khan3
1 Department of Medicine, Hackensack University Medical Center, Hackensack, New Jersey; 2Massey Cancer Center, Richmond, Virginia; 3Roswell Park Cancer Institute, Buffalo, New York
Latif and colleagues1 reported a case of erythroleukemia presenting as acute myeloid leukemia (AML) in a patient who was treated with a standard induction regimen. The patient experienced disease evolution and required treatment with salvage treatment, but expired due to disease progression. Erythroleukemia is a rare subtype of AML, which is categorized in the poor-risk group of AML (French-American-British [FAB] M6, or Di-Guglielmo’s disease <5%). Since the initial recognition, the diagnosis and classification of erythroleukemia has been significantly modified. The FAB classification in 1976 recognized erythroleukemia as M6; in 1985, the FAB classification defined erythroleukemia as leukemia with a major erythroid component of at least 50% and a myeloid component of approximately 30% of the nonerythroid cells.1,2 The World Health Organization (WHO) classifies erythroleukemia into 2 distinct variants, one with a combined erythroid and myeloid component and the other, which is pure erythroleukemia. Historically, the identification of erythroleukemia dates back to 1912, when it was initially recognized by Copelli as a hematologic disorder and named as erythematosus; in 1917 Di Guglielmo described eritroleucemia as proliferation of abnormal erythroid cells, myeloblasts, and megakaryocytes with the presence of immature forms of all 3 lineages in the peripheral blood. Di Guglielmo used the term eritroleucopiastrinaemia to demonstrate trilineage involvement and presented the idea that the abnormality was arising in the myeloid tissue with involvement of all 3 cytopoietic components.3 Moeschlin described the term erythroleukemia in 1940 and was followed by Dameshek and Baldini, who described the Di Guglielmo syndrome as propagation of 3 phases in the bone marrow: an erythremic phase, an erythromyeloblastic phase, and a myeloblastic phase analogous to AML.4
Erythroleukemia is a rare subtype of AML and represents 3–5% of adult AML cases; the incidence is rare in children. The median age at diagnosis is in the fifth to sixth decade of life.5,6 Another distinguishing feature of erythroleukemia is the predominant association of antecedent diagnosis of myelodysplastic syndrome along with multiple poor-risk chromosomal abnormalities identified on cytogenetic analysis.5,7 Erythroleukemia presents with a significant number of genomic aberrations, the common being abnormalities of chromosome 5 and/or 7 and complex karytoype abnormalities, which have been reported in most series.7,8 Aberration of chromosome 19 has also been reported in 1 study.9 The median survival varies from 4 to 14 months.6 The characteristic hematologic features of erythroleukemia include anemia, which is usually normochromic and normocytic, with severe anisocytosis.10 Nucleated red blood cells are preponderous, with the most immature and basophilic forms found in the largest numbers. Atypical nucleated red blood cells are also commonly seen, and reticulocytes are few, with a gradual decrease seen with disease progression.10 Leukocytes exhibit leukopenia along with thrombocytopenia as the common abnormalities. Reticulo-endothelial cells have also been observed in peripheral blood. The bone marrow examination exhibits an increase in red cell series and a decrease in the number of white cells.10 The nucleated red to white cell ratio is reversed, with a predominance of basophilic erythroblasts as well as proerythroblasts resulting in the appearance of arrested red cell maturation. The cells that stain positive for PAS, and the absence of staining of other lineage-specific markers, such as myeloperoxidase (MPO) and terminal transferase detection (TDT), are suggestive of erythroleukemia.7 The infiltration of these primitive red cells and reticulo-endothelial cells is observed in hematopoietic and extra-hematopoietic organs like the kidneys, adrenals, myocardium, lungs, pancreas, testes, uterus, larynx, trachea, and spleen.10
The standard induction therapy for AML continues to be based on anthracycline and cytarabine, which results in complete remission rates of approximately 70%, with a long-term survival of 30–40%; however, the prognosis for the high-risk group continues to be worse, with a long-term survival rate of less than 10%.11,12 Erythroleukemia is considered in the highrisk group and is associated with poor prognosis due to the common association with high-risk chromosomal abnormalities; it evolves in the background of dysplasia, with a median survival of 4–14 months. Additionally, the rarity of this leukemia makes it difficult for evaluation of treatment in randomized studies. Although the data are scarce for allogeneic transplantation in erythroleukemia due to the rarity of disease, allogeneic stem cell transplantation offers durable responses as compared to conventional chemotherapy alone. Anecdotal reports along with a study from the Royal Marsden Group presented outcomes of 27 patients undergoing allogeneic transplantation; 19 patients who underwent allogeneic transplantation achieved an overall survival rate of 66% at 2 years.13,14 The European Group for Blood and Marrow Transplantation (EBMT) reported the largest series of patients with erythroleukemia treated with autologous or allogeneic stem cell transplantation in first complete remission. For autologous transplantation, the 5-year leukemia-free survival was 26% ±5%, the relapse incidence was 70% ±6%, and the transplant-related mortality was 13% ±4%; for allogeneic transplantation, the 5-year leukemia-free survival was 57% ±5%, the relapse incidence was 21% ±5%, and transplant-related mortality was 27% ±5%.5
The outcome of patients with AML is heterogeneous and dependent on a number of risk factors. The important prognostic indicators in AML include karyotype, presence of antecedent hematologic disorders, age, and performance status. Among the new prognostic markers, the fms-tyrosine kinase (FLT3) accounts for the most frequent molecular mutations in AML.15 FLT3 is a transmembrane tyrosine kinase receptor that stimulates cell proliferation upon activation. FLT3 length mutation (FLT3-LM or FLT3-ITD for internal tandem duplication) is among the frequent genetic alterations in AML associated with prognostic implications. The frequency of FLT3-LM is 20–27% in adult AML and 10–16% in childhood AML.15 Studies with patients harboring this mutation have reported an estimated 2-year progressionfree survival of 20% and 4-year overall survival of 20%.16,17 Additionally, FLT3 also harbors mutations in the tyrosine kinase domains (FLT3-TKD mutations). Although the FLT3-TKD is less frequent than FLT3-ITD mutations, incidence of 5.8–7.7% have been described in reports. The prognosis of patients with the FLT3-TKD mutations is not associated with dismal outcomes.17 Ongoing research is delineating the impact of various types of FLT3 mutations on the outcomes of patients with AML.18 It has been observed that the incidence of FLT3-TKD is less frequent in erythroleukemia, which is attributed to an association with a complex aberrant karyotype.15 Erythroleukemia is a rare subtype of AML with a frequent association with aberrant karyotype and resultant aggressive disease course. Thus, it is a challenge to investigate and identify new treatments. Newer prognostic markers are being applied in AML along with erythroleukemia. Better understanding and identification of molecular markers offers new avenues for therapy. FLT3 inhibitors are already being applied in clinical trials with promising results, and clinical applications are eagerly awaited.19,20 Thus, it is likely that the future may hold promise for erythroleukemia in the lieu of molecular markers and targeted treatment. The current treatment is standard induction along with consolidation with allogeneic transplant, if available, which appears to provide better disease control than consolidation chemotherapy alone.
1. Bennett JM, Catovsky D, Daniel MT, et al. Proposals for the classification of the acute leukaemias. French-American-British (FAB) co-operative group. Br J Haematol. 1976;33:451-458.
2. Bennett JM, Catovsky D, Daniel MT, et al. Proposed revised criteria for the classification of acute myeloid leukemia. A report of the French-American-British Cooperative Group. Ann Intern Med. 1985;103:620-625.
3. Bain BJ. Di Guglielmo and his syndromes. Br J Haematol. 2003;120:939-943.
4. Dameshek W, Baldini M. The Di Guglielmo syndrome. Blood. 1958;13: 192-194.
5. Fouillard L, Labopin M, Gorin NC, et al. Hematopoietic stem cell transplantation for de novo erythroleukemia: a study of the European Group for Blood and Marrow Transplantation (EBMT). Blood. 2002;100:3135-3140.
6. Roggli VL, Saleem A. Erythroleukemia: a study of 15 cases and literature review. Cancer. 1982;49:101-108.
7. Santos FP, Faderl S, Garcia-Manero G, et al. Adult acute erythroleukemia: an analysis of 91 patients treated at a single institution. Leukemia. 2009;23: 2275-2280.
8. Nakamura H. Cytogenetic heterogeneity in erythroleukemia defined as M6 by the French-American-British (FAB) Cooperative Group criteria. Leukemia. 1989;3:305-309.
9. Cigudosa JC, Odero MD, Calasanz MJ, et al. De novo erythroleukemia chromosome features include multiple rearrangements, with special involvement of chromosomes 11 and 19. Genes Chromosomes Cancer. 2003;36:406-412.
10. Schwartz SO, Critchlow J. Erythremic myelosis (Dl Guglielmo’s disease); critical review with report of four cases, and comments on erythroleukemia. Blood. 1952;7:765-793.
11. Karp JE, Smith MA. The molecular pathogenesis of treatment-induced (secondary) leukemias: foundations for treatment and prevention. Semin Oncol. 1997;24:103-113.
12. Lowenberg B, Downing JR, Burnett A. Acute myeloid leukemia. N Engl J Med. 1999;341:1051-1062.
13. Kogawa K, Sekine I, Masuda T, et al. Bone marrow transplantation for erythroleukemia: a case report. Acta Paediatr Jpn. 1994;36:693-696.
14. Killick S, Matutes E, Powles RL, et al. Acute erythroid leukemia (M6): outcome of bone marrow transplantation. Leuk Lymphoma. 1999;35:99-107.
15. Bacher U, Haferlach C, Kern W, Haferlach T, Schnittger S. Prognostic relevance of FLT3-TKD mutations in AML: the combination matters—an analysis of 3082 patients. Blood. 2008;111:2527-2537.
16. Marcucci G, Maharry K, Whitman SP, et al. High expression levels of the ETS-related gene, ERG, predict adverse outcome and improve molecular risk based classification of cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B Study. J Clin Oncol. 2007;25:3337-3343.
17. Schlenk RF, Dohner K, Krauter J, et al. Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia. N Engl J Med. 2008;358: 1909-1918.
18. Loriaux MM, Levine RL, Tyner JW, et al. High-throughput sequence analysis of the tyrosine kinome in acute myeloid leukemia. Blood. 2008;111:4788-4796.
19. Pratz KW, Sato T, Murphy KM, Stine A, Rajkhowa T, Levis M. FLT3 mutant allelic burden and clinical status are predictive of response to FLT3 inhibitors in AML. Blood. 2010;115:1425-1432.
20. Pratz KW, Cortes J, Roboz GJ, et al. A pharmacodynamic study of the FLT3 inhibitor KW-2449 yields insight into the basis for clinical response. Blood. 2009;113:3938-3946.
- ASH Foundation
- Log in or create an account
- Publications
- Diversity Equity and Inclusion
- Global Initiatives
- Resources for Hematology Fellows
- American Society of Hematology
- Hematopoiesis Case Studies
Case Study: Prognostic Factors in Acute Lymphocytic Leukemia
- Agenda for Nematology Research
- Precision Medicine
- Genome Editing and Gene Therapy
- Immunologic Treatment
- Research Support and Funding
A 48-year-old female presented to the emergency department with severe headaches, dyspnea on exertion, and petechiae on the lower extremities. A CBC was drawn that showed the following: WBC=56 x10 3 /µL, Hgb=9.0 g/dL, Hct=23, MCV=97 fl, plt=15 x10 9 /µL, ANC=0.7x10 3 /µL. A bone marrow biopsy was performed and showed 90 percent lymphoid blasts. The blast population expressed CD19, CD20, CD22, CD10, CD34, TdT, and HLA-DR. Metaphase cytogenetics showed a normal female karyotype. FISH for bcr-abl was positive. The patient was subsequently diagnosed with pre-B acute lymphoblastic leukemia.
The abnormality of which gene is associated with a poor outcome in this patient?
- Ten-Eleven Translocation-2 (TET2)
- Isocitrate Dehydrogenase 1/2 ;(IDH1/IDH2)
Explanation
IKAROS ( IKZF1 ), a gene located in chromosome 7p12, is responsible for encoding a zinc finger-containing transcription factor called Ikaros that is essential for normal lymphoid development. Using genome-wide analysis, deletions involving IKZF1 were found in 84 percent of 304 patients with acute lymphoblastic leukemia (ALL). 1 Ikaros can occur in several splice variants and may explain some of the differential function of this transcription factor. The expression of certain spliced oncogenic Ikaros isoforms in bcr-abl-positive ALL may confer resistance to tyrosine kinase inhibitors (TKIs). 2 In a study of 83 patients with bcr-abl-positive ALL, a high frequency of IKZF1 deletions was observed (63 percent), mainly in exons 4 to 7 and exons 2 to 7. Patients who carry the IKZF1 deletions have inferior disease-free survival compared to those who have wild-type IKZF1 (10 months versus 32 months). 3
Ten-eleven translocation 2 ( TET2 ) is a gene located in chromosome 4q24. Mutations involving this gene can be found in varying frequencies in myeloid malignancies, including 10 percent in myelodysplastic syndromes, 50 percent in chronic myelomonocytic leukemia (50 percent), 20 percent in myeloproliferative neoplasms (20 percent) and 20 percent in secondary acute myeloid leukemias derived from these conditions. Recent studies suggest that TET2 may be important in epigenetic regulation. The exact prognostic value of this mutation remains unclear although some suggest that patients who have TET2 mutations have a good prognosis. 4-6
NOTCH1 is a gene that encodes for a transmembrane receptor that regulates normal T-cell development. This mutation has been detected in > 50 percent of both pediatric and adult T-ALL cases. FBXW7 encodes an E3 ubiquitin ligase responsible for negative regulation of NOTCH1 signaling. Mutations involving FBXW7 occur at a frequency of about 20 percent in ALL. Pediatric studies suggest the good prognostic value of NOTCH1 / FBXW7 mutations. The results in adult ALL patients are less clear, with conflicting results. 7-9
Isocitrate Dehydrogenase 1 ( IDH1 ) is a gene located in chromosome 2q33.3, while IDH2 is located in chromosome 15q26.1. These genes are responsible for encoding enzymes that catalyze the oxidative decarboxylation of isocitrate to α-ketoglutarates. Intact IDH activity is necessary for normal cellular protection from oxidative stress. These mutations were first described in low-grade gliomas/secondary glioblastomas and subsequently in acute myeloid leukemia. Some studies suggest that mutations involving this gene may confer poor prognosis in certain subsets, particularly in cytogenetically normal FMS-like tyrosine kinase-3 ( Flt-3 ), wild-type, and nucleophosmin-1 ( NPM-1 )-mutated AML. 10-12
- Mullighan CG, Miller CB, Radtke I, et al. BCR–ABL1 lymphoblastic leukaemia is characterized by the deletion of Ikaros . Nature. 2008;453:110-114.
- Iacobucci I, Lonetti A, Messa F, et al. Expression of spliced oncogenic Ikaros isoforms in Philadelphia-positive acute lymphoblastic leukemia patients treated with tyrosine kinase inhibitors: implications for a new mechanism of resistance . Blood. 2008;112:3847-3855.
- Martinelli G, Iacobucci I, Storlazzi CT, et al. IKZF1 (Ikaros) deletions in BCR-ABL1 –positive acute lymphoblastic leukemia are associated with short disease-free survival and high rate of cumulative incidence of relapse: a GIMEMA AL WP report . J Clin Oncol. 2009;27:5202-5207.
- Jankowska AM, Szpurka H, Tiu RV, et al. Loss of heterozygosity 4q24 and TET2 mutations associated with myelodysplastic/myeloproliferative neoplasms . Blood. 2009;113:6403-6410.
- Kosmider O, Gelsi-Boyer V, Cheok M, et al. TET2 mutation is an independent favorable prognostic factor in myelodysplastic syndromes (MDSs) . Blood. 2009;114:3285-3291.
- Tefferi A, Pardanani A, Lim KH, et al. TET2 mutations and their clinical correlates in polycythemia vera, essential thrombocythemia and myelofibrosis . Leukemia. 2009;23:905-911.
- Asnafi V, Buzyn A, Le Noir S, et al. NOTCH1/FBXW7 mutation identifies a large subgroup with favorable outcome in adult T-cell acute lymphoblastic leukemia (T-ALL): a group for research on adult acute lymphoblastic leukemia (GRAALL) study . Blood. 2009;113:3918-3924.
- Park MJ, Taki T, Oda M, et al. FBXW7 and NOTCH1 mutations in childhood T cell acute lymphoblastic leukaemia and T cell non-Hodgkin lymphoma . Br J Haematol. 2009;145:198-206.
- Mansour MR, Sulis ML, Duke V, et al. Prognostic implications of NOTCH1 and FBXW7 mutations in adults with T-cell acute lymphoblastic leukemia treated on the MRC UKALLXII/ECOG E2993 protocol . J Clin Oncol. 2009;27:4352-4356.
- Tefferi A, Lasho TL, Abdel-Wahab O, et al. IDH1 and IDH2 mutation studies in 1473 patients with chronic-, fibrotic- or blast-phase essential thrombocythemia, polycythemia vera or myelofibrosis . Leukemia. 2010;24:1302-1309.
- Abbas S, Lugthart S, Kavelaars FG, et al. Acquired mutations in the genes encoding IDH1 and IDH2 both are recurrent aberrations in acute myeloid leukemia (AML): prevalence and prognostic value . Blood. 2010. [Epub ahead of print]
- Paschka P, Schlenk RF, Gaidzik VI, et al. IDH1 and IDH2 mutations are frequent genetic alterations in acute myeloid leukemia and confer adverse prognosis in cytogenetically normal acute myeloid leukemia with NPM1 mutation without FLT3 internal tandem duplication . J Clin Oncol. 2010;28:3636-3643.
Case study submitted by Sanjay R. Mohan, MD, Cleveland Clinic Taussig Cancer Institute.
American Society of Hematology. (1). Case Study: Prognostic Factors in Acute Lymphocytic Leukemia. Retrieved from https://www.hematology.org/education/trainees/fellows/case-studies/acute-lymphocytic-leukemia .
American Society of Hematology. "Case Study: Prognostic Factors in Acute Lymphocytic Leukemia." Hematology.org. https://www.hematology.org/education/trainees/fellows/case-studies/acute-lymphocytic-leukemia (label-accessed August 18, 2024).
"American Society of Hematology." Case Study: Prognostic Factors in Acute Lymphocytic Leukemia, 18 Aug. 2024 , https://www.hematology.org/education/trainees/fellows/case-studies/acute-lymphocytic-leukemia .
Citation Manager Formats
- Biomarker-Driven Lung Cancer
- HER2-Positive Breast Cancer
- Chronic Lymphocytic Leukemia
- Small Cell Lung Cancer
- Renal Cell Carcinoma

- CONFERENCES
- PUBLICATIONS
- CASE-BASED ROUNDTABLE
Case-Based Overview: Newly Diagnosed Acute Myeloid Leukemia
Naval G. Daver, MD: This is the case of a 64-year-old patient with newly diagnosed acute myeloid leukemia. The patient has significant comorbidities, including an elevated BMI [body mass index], signifying a significant obesity, as well as underlying pneumonia and prior history of high blood pressure and diabetes. These are all comorbidities that make us start thinking about the optimal therapeutic choice for this patient. It’s also very important to confirm the diagnosis for this patient, and the bone marrow that has been done does show a diagnosis of acute myeloid leukemia with more than 20% blast, which meets the WHO [World Health Organization] criteria.
The main things that I’m thinking about at this time for this patient are, number 1, stabilizing him. The CT [computed tomography] scan does show that he has an active infiltrate concerning for an infectious pneumonia. So, I would probably start the patient on some oral or IV [intravenous] antibiotics, depending on his clinical signs and symptoms. And then most importantly, try to rush the molecular and chromosome testing to try and identify the optimal frontline therapy.
At this time, I’m probably not inclined to go with intensive induction therapy for this patient. This is not just because of his agehe is 64, which is still a patient that could be eligible for intensive induction with cytarabine, anthracycline—but more so because of his significant comorbidities: the elevated BMI, the ongoing pneumonia, the blood pressure and diabetes issues. I’m thinking of going with lower intensity therapies such as hypomethylating agent or low-dose cytarabine with venetoclax, or potentially targeted therapy-based combinations if we find a targetable mutation such as FLT3 or IDH1 , IDH2 mutations.
Historically for such a patient we would have considered standard induction therapy, which is a combination of cytarabine anthracycline. There are many different ways this induction therapy can be given. The most common is what we call “3 plus 7,” which is 3 days of the anthracyclineeither idarubicin or daunorubicin—and 7 days of the cytarabine, either 100 or 200 per meter square. There are variations, some of which have shown higher response rates, such as FLAG-IDA [fludarabine, cytarabine, idarubicin and filgrastim], CLIA [idarubicin plus high‐dose cytarabine + clofarabine/cladribine], and others. So that would probably be the standard therapy that we’ve used for acute myeloid leukemia for about 40 years now.
If this patient was diagnosed 4 or 5 years ago, for example, that would be the most likely treatment approach. We also did have lower intensity therapies such as Vidaza [azacitidine], decitabine. These have been available now for the last 12 or 13 years. The problem was that the response rates with Vidaza or decitabine as single-agent were only about 20% to 28%, which is much lower than the 65% to 75% you could get with 7 + 3. In most patients we would try to push to give them 7 + 3 unless, of course, they were extremely sick in the ICU [intensive care unit] or had really severe comorbidities.
But now with the advent of the new combinations such as azacitidine or decitabine with venetoclax added, we’re seeing the response rates are going up to about 70% to 75%. Now, we actually don’t necessarily push for the traditional 7 + 3 induction if we feel that the patient may not tolerate it but go for the azacitidine with venetoclax type of approach instead.
Transcript edited for clarity.
Case: A Male With Rapidly Progressing Acute Myeloid Leukemia
A 64-year-old male presented with a 2-week history of subjective fever, fatigue, shortness of breath, dizziness, and cough
- PE: Temperature 99.1 o F, pallor of the conjunctiva, multiple ecchymosis on upper and lower extremities
- PMH: DM controlled on metformin, hypertension, BMI >35, recent history of pneumonia treated with oral antibiotics
Diagnostic Work- Up
- WBC: 2.3 x 10 3 /µL, RBC: 3.1212 x 10 6 /µL, Hb: 9.3 g/dL, Ht: 23.1%, Plt: 83 x 10 3 /µL, LDH: 275 U/L, blasts: 36%, absolute neutrophil count: 320 cells/µL, PT: 16.1s,
- Few auer rods noted on bone marrow aspiration
- Diagnosed with AML with 43% blasts on pathology evaluation, flow-cytometry confirms AML
- Molecular panel and cytogenic testing pending and RUSH requested
- Chest CT revealed patchy consolidation in the left lower lung lobe with ill-defined nodules
- EKG and Echocardiogram unremarkable
- Started on prophylactic voriconazole, cefpodoxime, and valacyclovir
- Patient was started at this time on azacitidine and venetoclax; Azacitidine 75mg/m 2 Days 1-7 and Venetoclax Days 1-28. Venetoclax dose was 100mg with voriconazole.
- Was admitted for tumor lysis monitoring and hydration. Tolerated cycle 1 well. continue until disease progression or unacceptable toxicity
- Day 28 post-treatment bone marrow aspirate revealed low percent residual blasts (3% blasts by flow) with hypocellular BM (5-10% cellularity) and ANC 0.3, platelets 23K
- Venetoclax was interrupted at this time. Labs checked 2-3 times per week outpatient. Within 12 days after venetoclax interruption ANC>0.5 and platelets>50K.
- Cycle 2 started outpatient with standard dose azacitidine and venetoclax reduced to 14-21 days
- Patient subsequently developed pneumonia, treated with oral antibiotics
- Patient will continue routine bone marrow biopsies after cycle 4, and every 6 months thereafter or if disease progression is suspected

Frontline Acalabrutinib/BR Regimen Improves PFS in MCL
Acalabrutinib plus bendamustine and rituximab led to a 27% reduction in the risk of disease progression or death in the frontline setting for older patients with mantle cell lymphoma.

DeZern Discusses Incorporating Current Options for Managing Anemia in Low-Risk MDS
During a Case-Based Roundtable® event, Amy DeZern, MD, MHS, discussed dosing approaches for managing anemia with luspatercept in patients with low-risk myelodysplastic syndrome.

FDA Approves Improved Denileukin Diftitox in Cutaneous T-Cell Lymphoma
Following voluntary withdrawal in 2014, denileukin diftitox is now available again for the treatment of patients with cutaneous T-cell lymphoma who have received at least 1 prior systemic therapy.

FDA Rejects Iomab-B BLA Application, Requests New Randomized Study
The FDA has rejected the SIERRA study data for Iomab-B’s biologics license application filing due to insufficient evidence of overall survival improvement.
Alemtuzumab Wins FDA Orphan Drug Designation as Part of ALL CAR T Therapy
The FDA has granted an orphan drug designation to alemtuzumab, a chimeric antigen receptor T-cell therapy for relapsed/refractory B-cell acute lymphoblastic leukemia.
2 Commerce Drive Cranbury, NJ 08512
609-716-7777

- Alzheimer's disease & dementia
- Arthritis & Rheumatism
- Attention deficit disorders
- Autism spectrum disorders
- Biomedical technology
- Diseases, Conditions, Syndromes
- Endocrinology & Metabolism
- Gastroenterology
- Gerontology & Geriatrics
- Health informatics
- Inflammatory disorders
- Medical economics
- Medical research
- Medications
- Neuroscience
- Obstetrics & gynaecology
- Oncology & Cancer
- Ophthalmology
- Overweight & Obesity
- Parkinson's & Movement disorders
- Psychology & Psychiatry
- Radiology & Imaging
- Sleep disorders
- Sports medicine & Kinesiology
- Vaccination
- Breast cancer
- Cardiovascular disease
- Chronic obstructive pulmonary disease
- Colon cancer
- Coronary artery disease
- Heart attack
- Heart disease
- High blood pressure
- Kidney disease
- Lung cancer
- Multiple sclerosis
- Myocardial infarction
- Ovarian cancer
- Post traumatic stress disorder
- Rheumatoid arthritis
- Schizophrenia
- Skin cancer
- Type 2 diabetes
- Full List »
share this!
August 14, 2024
This article has been reviewed according to Science X's editorial process and policies . Editors have highlighted the following attributes while ensuring the content's credibility:
fact-checked
peer-reviewed publication
trusted source
New research poised to transform approach to diagnosing and treating acute leukemia in children
by St. Jude Children's Research Hospital
. DOI: 10.1038/s41586-024-07807-0")
Researchers at Children's Hospital of Philadelphia (CHOP), St. Jude Children's Research Hospital (St. Jude) and the Children's Oncology Group (COG) today announced a significant paradigm shift in the understanding of T-lineage acute lymphoblastic leukemia (T-ALL), an aggressive and high-risk form of cancer, to one frequently driven by genetic changes in non-coding portions of our DNA.
The collaborative study was published today in the journal Nature .
Many children, adolescents, and young adult patients with T-ALL traditionally respond well to initial treatment . However, patients who relapse or have treatment-resistant disease often face a dire prognosis. Given the aggressive nature and rapid progression of the disease, and limited understanding of the genetic basis of T-ALL, researchers saw an urgent need for new and effective approaches to diagnosis and treatment.
"This paper is the first to transcend previous barriers and comprehensively profile the whole genome, uncovering critical insights in more than 1,300 children, adolescents and young adults with T-ALL," said David T. Teachey, MD, an attending physician, Director of Clinical Research at the Center for Childhood Cancer Research at CHOP and Chair of the Acute Lymphoblastic Leukemia disease committee in the COG.
"These findings are a significant clinical advancement, as the goal in treating T-ALL is to prevent relapse, which requires identifying the patients most at risk. This data now makes it possible to risk stratify patients with T-cell leukemia, identifying those with a high-risk of relapsing so we can treat them with newer or alternative medicines."
Prior studies were unable to identify important genetic changes in T-ALL, as they focused on the coding genome, the part of DNA that encodes proteins, the building blocks of cells. However, only 1% of DNA is coding, while the other 99% is termed non-coding.
Once considered useless, scientists now recognize that the non-coding region plays a key role in regulating biological processes. It signals the cell when to produce certain proteins, like a crossing guard aiding people to safely cross the street.
In this case, researchers studied more than 1,300 patients treated on the COG AALL0434 clinical trial and sequenced both the tumor and non-tumor genomes of each patient. While the researchers previously suspected that non-coding DNA in T-ALL played an important role, this study's findings are the first ever to establish that at a large scale.
The study found that approximately 60% of the genetic changes driving T-ALL cancer cells are non-coding changes. This fundamentally alters the way researchers think about T-ALL, offering a better understanding of disease biology. This leads to innovative treatments, including new immunotherapies developed at CHOP and St. Jude.
Traditionally, patients with T-All have been categorized by risk based on their therapy response and immunophenotype, which profiles cell surface proteins as part of the diagnostic workup. While cell surface protein expression helps define T-ALL subtypes, it hasn't proven effective in consistently identifying which patients have a good prognosis.
The new comprehensive data revealed why, strongly suggesting that a genomic approach replace the current immunophenotypic classification. As a result, the researchers developed models that incorporate genetics and response to treatment to risk stratify patients with T-ALL accurately and are currently in the process of validating results using patient samples from the next COG trial of T-ALL.
"It was striking how abundant these non-coding changes were and how many of them were enhancer perturbation events, whether it was hijacking or co-option of an existing enhancer, or changes that generated a new enhancer," said Charles Mullighan, MBBS, MD, St. Jude Children's Research Hospital, Comprehensive Cancer Center deputy director and Department of Pathology member.
"We now have a much stronger framework to take these alterations back to the lab and say now we've got better information to build the right models to understand the biology, and then to test therapy. We have very clear information that these are the sorts of alterations that people need to focus on to build a diagnostic test."
Researchers were able to classify T-ALL into 15 subtypes with distinct gene expression and genomic drivers, including previously undefined subtypes. They refined the classification of known subtypes and showed that driver lesions, other genetic changes, and the original cell type work together to define the genomic subtype and the clinical and biological characteristics of a condition.
They also observed a significant link between the type of gene alterations and outcomes in T-ALL. This new observation shows it is not only which gene is altered in the cancer cells , but also how it is altered, that helps define prognosis and chance of a cure.
"Future research must continue to determine broader applications for this approach," said Teachey. "These findings offer a strong a roadmap for improving patient outcomes and curing more children and adults with T-ALL."
Explore further
Feedback to editors

Researchers develop new chemical method to enhance drug discovery
Aug 17, 2024

Rare diseases point to connections between metabolism and immunity

Researchers discover novel nanoparticles in blood with potential to transform cancer diagnosis

Research shows how to reduce inappropriate IV use by more than a third
Aug 16, 2024

Now that mpox is a global health emergency, will it trigger another pandemic?

Knocking out one key gene leads to autistic traits, mouse study shows

Study: Rare cancer patients nearly three times more likely to develop anxiety and depression than common cancer patients

Intervention for cleaning shared health care equipment could significantly reduce health care–associated infections

Lip reading activates brain regions similar to real speech, researchers show

Parents' excessive smartphone use could harm children's mental health
Related stories.

Scientists identify genes linked to relapse in the most common form of childhood leukemia
Aug 12, 2024

Study identifies high-risk type of childhood acute leukemia and potential treatment strategy
Jun 26, 2024

Genetic signatures provide prognostic information in colorectal cancer
Aug 7, 2024

Age impacts pharmacogenomics and treatment outcomes for most common form of leukemia
Aug 6, 2024

Unraveling the roles of non-coding DNA explains childhood cancer's resistance to chemotherapy
May 1, 2024

Risk-directed childhood leukemia treatment takes a step forward
Apr 12, 2021
Recommended for you

Can a mouthwash-based test help predict head and neck cancer recurrence?
Aug 15, 2024

AI shows greater efficiency in detecting early signs of bowel cancer

First successful treatment of pediatric high-risk refractory neuroblastoma with PARP inhibition

Study unveils impact of cardiovascular risk factors on genetic predisposition to heart disease
Let us know if there is a problem with our content.
Use this form if you have come across a typo, inaccuracy or would like to send an edit request for the content on this page. For general inquiries, please use our contact form . For general feedback, use the public comments section below (please adhere to guidelines ).
Please select the most appropriate category to facilitate processing of your request
Thank you for taking time to provide your feedback to the editors.
Your feedback is important to us. However, we do not guarantee individual replies due to the high volume of messages.
E-mail the story
Your email address is used only to let the recipient know who sent the email. Neither your address nor the recipient's address will be used for any other purpose. The information you enter will appear in your e-mail message and is not retained by Medical Xpress in any form.
Newsletter sign up
Get weekly and/or daily updates delivered to your inbox. You can unsubscribe at any time and we'll never share your details to third parties.
More information Privacy policy
Donate and enjoy an ad-free experience
We keep our content available to everyone. Consider supporting Science X's mission by getting a premium account.
E-mail newsletter
- Campus Directory
- Current Students
- Faculty & Staff

Acute Lymphocytic Leukemia Case Study
Leukemia is a cancer of white blood cells. In acute leukemia, the abnormal cells divide rapidly, quickly overtaking functional white and red blood cells. The most common form of cancer in children 0-14 years of age is acute lymphocytic leukemia (ALL). The survival rate in children has improved more than 50% in the last half century. Currently, there is a 65.3% overall survival rate; in children under 5 the survival rate increases to 90.4%. Come experience the cancer journey with 6-year-old Noah.
Module 6: Acute Lymphocytic Leukemia
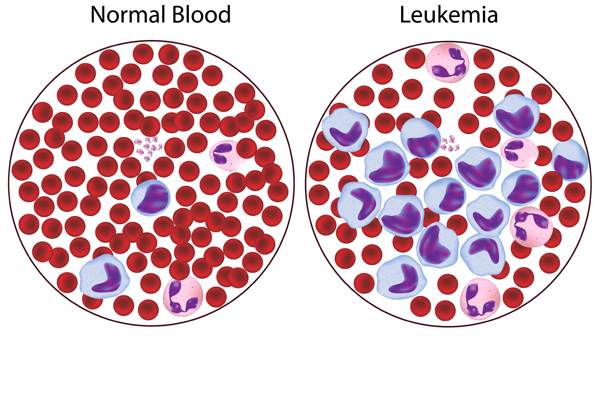
Noah, 6 years old, was brought back to his pediatrician three weeks following a streptococcal throat infection...
Leukemia - Page 1
Upon receiving the results, the physician informed the stunned mother that her child had...
Leukemia - Page 2
A spinal tap was ordered to see if the leukemic cells had crossed the blood/brain barrier...
Leukemia - Page 3
A sputum culture was obtained and sent to the lab for gram stain, culture, and sensitivities...
Leukemia - Page 4
Noah was now in remission. Two weeks after achieving...
Leukemia - Page 5

Case Summary
Summary of the Case
Leukemia - Summary

Answers to Case Questions
Leukemia - Answers

Professionals
Health Professionals Introduced in Case
Leukemia - Professionals
Additional Links
Optional Links to Explore Further
Leukemia - Links

An official website of the United States government
The .gov means it’s official. Federal government websites often end in .gov or .mil. Before sharing sensitive information, make sure you’re on a federal government site.
The site is secure. The https:// ensures that you are connecting to the official website and that any information you provide is encrypted and transmitted securely.
- Publications
- Account settings
Preview improvements coming to the PMC website in October 2024. Learn More or Try it out now .
- Advanced Search
- Journal List
- BMJ Case Rep
Case Report
B-acute lymphoblastic leukaemia.
A 13-year-old boy presented with fever, skeletal pain, polydipsia, polyuria and multiple osteolytic lesions in pelvic bones and upper femur. There was no organomegaly or lymphadenopathy. His serum calcium levels were raised. Peripheral blood film examination was normal. Bone marrow showed presence of blast cells. Flowcytometry indicated B-acute lymphoblastic leukaemia (B-ALL). Hypercalcaemia and osteolytic lesions are rare presentations of B-ALL. This should be kept as a differential if a child presents with unexplained skeletal pain with lytic lesions.
Hypercalcaemia and osteolytic bone lesions are rare presentations of B-acute lymphoblastic leukaemia (B-ALL) in contrast to their high incidence in some other lymphoid malignancies such as myeloma, Langerhan cell histiocytosis (LCH) and T-cell lymphoblastic leukaemia/lymphoma.
We think this presentation of leukaemia needs to be reported so that B-ALL can be kept in differential whenever a child presents with skeletal pain and multiple osteolytic lesions with normal peripheral blood picture.
Case presentation
A 13-year-old boy presented with malaise and fever associated with anorexia, pain in both lower limbs, polyuria and polydipsia, on and off for 3 months.
On examination he had pallor and bone tenderness. There was no organomegaly or lymphadenopathy.
Investigations
The boy was referred to the radiology department. X-rays revealed multiple lytic lesions in the pelvis and upper femur of both lower limbs ( figure 1 ). A provisional diagnosis of LCH was made.

X-ray pelvis showing multiple lytic lesions.
Biochemical parameters were: alkaline phosphatase 156 IU/L, lactate dehydrogenase 1650 IU/L, uric acid 5 mg/dL, albumin 3.4 g/dL and serum calcium 14 mg/dL.
He was referred to the haematology department for further work up. His haemogram revealed haemoglobin 9.1 g/dL, total leucocyte count 6300/mm 3 , differential leucocyte count: polymorphs 69%, lymphocytes 22%, eosinophil 2% and monocytes 7%.
Bone marrow aspiration showed 92% blast cells with high nuclear cytoplasmic ratio, fine nuclear chromatin and inconspicuous nucleoli ( figure 2 ). Hence, diagnosis of ALL was made. Fine-needle aspiration cytology from the lytic lesions also showed similar blast cells ( figure 3 ).

Bone marrow aspirate showing blast cells (Leishman stain ×100).

Fine-needle aspiration cytology from lytic lesions showing similar blast cells (Leishman stain ×40).
Flowcytometry of bone marrow aspirate showed positivity for CD34, CD10, CD19, CD20 and negativity for CD7, CD2, CD3, CD13 and CD33.
As a result, confirmatory diagnosis of B-ALL was made.
Differential diagnosis
Based on X-ray findings a differential diagnosis of LCH was made; however, presence of blast cells on bone marrow aspirate as well flowcytometry confirmed B-ALL as the diagnosis.
The patient was treated as per conventional protocol for B-ALL.
Outcome and follow-up
The patient has achieved complete remission but his bone lesions are still persisting. His serum calcium levels have normalised.
Acute lymphoblastic leukaemia (ALL) is the most common malignancy in the paediatric age group. 1 Symptoms of ALL include anaemia, fever, bleeding tendency and fatigue. 2 Hypercalcaemia and osteolytic lesions are rare in B-ALL in contrast to their incidence in some other lymphoid malignancies like adult T-cell leukaemia/lymphoma, myeloma and LCH. 2–4 However, in our case, due to the patient’s young age, myeloma was ruled out. Owing to lytic bone lesions the most likely diagnosis was LCH (eosinophilic granuloma), but bone marrow aspiration revealed the presence of blasts which on immunophenotyping by flowcytometry indicated B-ALL.
Lytic bone lesions with hypercalcaemia can be the initial signs even in the absence of blasts from peripheral blood smears. Hence in such cases bone marrow aspiration is mandatory ( table 1 ). Hypercalcemia in such cases is due to TNF (alpha and beta ), IL-2, IL-6, TGF beta, 1-25(OH)2 and direct invasion by the tumor cells or due to PTHrP and PGE2 secretion by the tumor cells. 4–7
Table 1
Different types of bony lesions in acute lymphoblastic leukaemia/lymphoma
| Author | Case number | Age/sex | Presenting symptoms | PBS/BMA* | Radiological findings |
|---|---|---|---|---|---|
| Shahnazi | Case 1 | 4 year/F | Back pain | No blasts in PBS; BMA revealed blasts | Multiple dorsolumbar collapsed vertebrae |
| Case 2 | 4 year/F | Pallor, weakness, wrist tenderness | PBS and BMA had blasts | Wrist X-ray revealed metaphyseal lucent band in radial and ulnar metaphyses and lucent bony lesions of the first and third metacarpal | |
| Case 3 | 8 year/M | Cough and musculoskeletal pain | Blasts in PBS and BMA | CXR reveals permeative bone lesion in the right humerus | |
| Case 4 | 8 year/M | Fever, back pain and limping | Blasts in PBS and BMA | Thoracolumbar AP and Lat X-rays revealed multiple collapsed vertebrae, knee X-ray revealed hypodensity around the knee joint with metaphyseal translucency | |
| Case 5 | 6 year/F | Pallor, weakness, pain in knee joint and elbow | Blasts in PBS and BMA | Periosteal reaction of radial proximal metaphysis with distal metaphyseal translucency (arrow); AP X-ray of knee joints revealed metaphyseal translucency in distal femurs and proximal tibiae | |
| Case 6 | 9 year/M | Groin pain and limping | Blasts in PBS and BMA | Reduced height of femoral epiphysis with osteochondral fracture on the left side due to avascular necrosis | |
| Case 7 | 13 year/F | Pallor, fever, headache and bone pain | Blasts in PBS and BMA | Skull X-ray revealed multiple lytic lesions; lumbosacral AP X-ray revealed permeative bony lesions in iliac bones with reduced vertebral height of L5; knee X-ray revealed reduced bone density with permeative appearance; CXR revealed permeative bony lesions in bilateral scapular bones | |
| Sirelkhatim | Case 8 | 17 year/M | Fever and backache | Blasts in PBS and BMA | Diffuse osteolytic lesions of lumbosacral area and pelvis |
| Case 9 | 12 year/F | Compression fracture of thoracic vertebrae | Blasts only on BMA | Osteolytic lesions in thoracic vertebrae and femur | |
| Case 10 | 7 year/M | Pain in lumbosacral area | Blasts only on BMA | Dorsal MRI showed atypical changes on MRI | |
| Chaudhary | Case 11 | 3 year/F | Knee pain and swelling | No blasts on PBS and BMA showed occasional abnormal cells | Expansile lytic lesion at the right supracondylar region and multiple lytic areas in the diaphyseal and metaphyseal regions of both femora and tibia |
*PBS is peripheral blood smear and BMA is bone marrow aspirate.
Our patient presented with multiple osteolytic lesions with hypercalcaemia and a normal total leucocyte count without any blasts in peripheral blood. It is not uncommon to find osteolytic lesions without circulating blasts in peripheral blood in patients of ALL ( box 1 ). Many such cases have been reported in the literature. 1 3 5 9 Therefore, ALL should always be kept as a differential in any child having multiple osteolytic lesions and hypercalcaemia even in the presence of normal peripheral blood findings.
Learning points
- Unexplained skeletal pain in children can have multiple aetiologies like trauma, infection and, rarely, leukaemia. So once trauma and infection are ruled out, leukaemia should be considered in differential.
- Lymphoblastic leukaemia should be considered in differential diagnosis whenever a child presents with osteolytic lesions and hypercalcaemia with normal peripheral blood film.
Factors associated with hypercalcaemia of malignancy 8
TNF-α, TNF-β
IL-1α, IL-1β, IL-6
TGF-α, TGF-β
Ectopic PTH
1,25 dihydroxyvitamin D
PG-E1, PG-E2
MIP-1α
Lymphotoxin
IL, interleukin; MIP, macrophage inflammatory protein; M-CSF, macrophage colony-stimulating factor; PG, prostaglandin; PTH, parathyroid hormone; PTHrP, parathyroid hormone-related peptide; RANKL, receptor activator of nuclear factor κB, ligand/osteoprotegerin system; TGF, transforming growth factor; TNF, tumour necrosis factor.
Contributors: RK undertook manuscript preparation, analysis, data collection and will act as guarantor. AK, MJ and USS helped in manuscript editing and manuscript review.
Competing interests: None.
Patient consent: Obtained.
Provenance and peer review : Not commissioned; externally peer reviewed.
- Open access
- Published: 16 August 2024
Exploring the lived experience of mothers of children with leukemia: a qualitative study from Iran
- Fatemeh Shaygani 1 , 2 ,
- Katayoun Jalali 3 ,
- Hana Javanmardi Fard 2 ,
- Zahra Afrasiabi 4 &
- Milad Ahmadi Marzaleh 5
BMC Women's Health volume 24 , Article number: 457 ( 2024 ) Cite this article
22 Accesses
Metrics details
Leukemia, as one of the most common pediatric cancers, has negatively affected many children around the world. Parents often experience increased feeling of distress shortly after being informed about their child’s diagnosis. The distress experienced by parents can adversely affect various aspects of their life. This study aimed to develop an understanding of the lived experience of the mothers whose children suffer from leukemia in Shiraz, Iran.
This phenomenological study was performed from April to August 2023, and 10 people were selected as participants by purposive sampling. In-depth and semi-structured interviews were performed for collecting the data.
The participants’ lived experiences during their children's leukemia were classified into five main categories, namely behavioral problems, spiritual issues, psychological problems, issues related to treatment, and economic matters.
Knowing the experiences of parents, especially mothers, in managing and planning for the care of these children seems essential.
Peer Review reports
According to statistics, it has been estimated that over 28 million cases of cancer will exist in 2040, which will contribute to a significant global burden of this important health issue [ 1 ]. Around 9.6 million people lost their lives due to this cancer in 2018, accounting for one in every six people dying from cancer-related issues [ 2 ]. It is also considered as one of the major causes of death worldwide among children and adolescents (aged 0–19 years old) [ 3 ], and yearly almost 400,000 children and adolescents develop cancer all over the world [ 4 , 5 ]. Based on the statistics, a growing number of individuals who have survived childhood and adolescent cancer (SCAC) is observed globally as advancements in cancer treatments, and supportive care is being improved [ 6 , 7 ].
Several studies conducted in the pediatrics field have generally focused on the negative psychosocial outcomes among both children with cancer and their family members [ 8 , 9 ].
According to the reports, parents often experience increased feeling of anxiety and depression shortly after being informed about their child’s diagnosis [ 10 , 11 ]. The prevalence of clinically relevant anxiety and depression among parents of children with cancer is as high as 74% and 46%, respectively [ 9 , 12 ].
The distress experienced by parents can adversely affect various aspects of their life such as quality of life, family dynamics, and marital satisfaction [ 8 ]. Nevertheless, the financial burden of cancer on families extends beyond medical expenses [ 13 ]. There are various additional costs that contribute to the overall burden, including direct costs such as medical care; indirect costs such as the loss of resources and opportunities; and psychosocial costs [ 13 , 14 , 15 , 16 ]. The psychosocial costs encompass intangible aspects associated with cancer, such as pain and suffering, and the impact on individuals' overall well-being [ 15 ].
Leukemia as the most prevalent cancer diagnosis in pediatric cases, which carries higher treatment costs compared to other types of pediatric cancers [ 17 , 18 ], is highly discussed in different research projects. To the best of our knowledge, there was no qualitative study in Iran assessing the lived experiences of parents of children with Leukemia, after the Coronavirus disease in 2019 pandemic. Therefore, this research was performed to explore the mothers’ experiences in Shiraz, the capital city of Fars and the fourth largest and the fourth most populated province in Iran.
Study design and data collection
In this qualitative study, which was conducted between April and August 2023 in Shiraz, Iran, a phenomenological approach was employed to explore the lived experience of mothers of children with leukemia. Since the chronic leukemia is rare among this group, there were only patients with acute leukemia, both Acute Lymphocytic Leukemia (ALL) and Acute Myelogenous Leukemia (AML). Additionally, we first included both types of ALL and AML patients’ mothers in the study to see if there is a remarkable difference in the lived experience of them according to the study’s result, we divide these groups. Data collection was accomplished using deep and semi-structured interviews with mothers who referred to the pediatric and adolescent cancer department of Imam Reza specialty and subspecialty clinic, which is the only center in Shiraz city where both inpatients and outpatients receive cancer-related medical services for at least once. It should be mentioned that all of the patients in this center were during the treatment period.
Mothers were selected using purposive sampling and recruited to be included in the study, just after receiving information about the objectives of the research and declaring their tendency for participation. We preferred to conduct in-person interviews, but whenever it was not possible, we replaced them with phone interviews. The written informed consent form was obtained from each participant in face-to-face interviews and in terms of phone-interviews; their tendency was verbally expressed and recorded in the audio file. Those mothers who were not willing to participate in the present study were excluded.
For conducting interviews, an initial interview guide was provided according to the literature review and the agreement among the professors and researchers of this field. Then, a pilot study was accomplished; conducting 2–3 interviews based on the initial interview guide and then the final version of interview guide was created to start the main research according to that. This guide included some questions, such as: "What experiences did you have when you realized that your child is a Leukemia patient?", "What challenges did you face when you realized that your child is a Leukemia patient?", and follow-up/probing questions such as "How?", "Why?", and "May you explain more…?”.
The interviews were conducted in a quiet place and at the time desired by the interviewee. To avoid possible problems or interruption in recording the voice of the interviewees, the interviewers recorded the session by two tape recorders.
Data analysis
Data collection and analysis were performed simultaneously. Recorded audio files were precisely transcribed by the interviewers. The researchers also took notes during the interview. After conducting each interview, its transcript was firstly written down and reviewed several times to get a general understanding of it. An interpretative summary was written for each interview transcript, and an attempt was made to understand and extract its meanings. To analyze the interviews, the Smith method [ 19 ] and MAXQDA software version 10 were used. The Smith method has six steps including: 1. Reading and re-reading the text, 2. Initial note taking, 3. Developing emerging codes, 4. Searching for any connection between codes, 5. Moving to a new code, and 6. Searching for final themes and sub-themes. In this study, we continued to conduct interviews until no new code was extracted and data saturation was achieved. The study finished with 10 interviews, which lasted from 45 to 100 min, and the average time was 60 min. Finally, review of literature was done after data analysis.
Trustworthiness
In order to ensure rigor in this qualitative study, we followed Guba and Lincoln’s method [ 20 ] following four standards: credibility, confirm ability, dependability, and transferability. An extreme variation sampling was considered in this study. Member checks with participants were performed during data collection and analysis, which mitigated the risk of misunderstanding and gave an opportunity to participants to check the accuracy and clarity of their experiences and make a change if needed. The collective opinions of the research team were also included in all stages of data analysis, and all the study steps were recorded with details. Furthermore, themes, sub-themes, and all codes were used to maintain the participants’ experiences and improve dependability.
Ethical considerations
The study protocol was approved by ethics committee of Shiraz University of Medical Sciences, with the code of IR.SUMS.NUMIMG.REC.1402.051. All methods were carried out in accordance with relevant guidelines and regulations or the Declaration of Helsinki. All participants were provided with sufficient information about the objectives of the study, and a written or verbal informed consent was obtained before beginning each interview. Interviewees were assured that the interviews would be confidential and audio files stored anonymously; also, they were assured that they could withdraw from the study at any stages.
In this study, the mean age of the participants was 32.9 years, with a range of 28 to 42 years. The demographic characteristics of the participants are shown in Table 1 .
After analyzing the interviews, we extracted 223 codes. According to our results, we did not observe a significant difference among the lived experience of mothers of children with ALL or AML types of leukemia, so we preferred to not divide the subjects according to these two types of leukemia and their lived experience included five main themes: behavioral problems, spiritual problems, psychological problems, challenges related to treatment, and economic tensions. Table 2 presents the sub-themes for each main theme.
The first theme extracted from the data analysis was behavioral problems, which included the sub-themes of disconnection from others, desire to be alone, silence, fear of pity and judgment, impatience, obsessive behavior, dependence on virtual space, aggression, and stubbornness. Having a child diagnosed with leukemia had caused most of the mothers to limit or cut off their communication with others and tend to be alone and silent. Many of these mothers reported impatience and fear of others' pity and judgment in this situation. Also, due to isolation, they spent more time in virtual space. In addition, according to their words, they had become aggressive and stubborn.
Mother 1 described her lack of communication with others and aggression as follows: “When we found out about my son's illness, I couldn't talk to anyone at all. If someone wanted to say hello, I tried not to talk to him/her because I was not in a good mood at all. We were very tense, I was nervous, I had become very aggressive, I was shouting, and I had completely lost my spirit.”
Mother 2 talked about her desire to be alone and silent and was afraid of other people's judgment like this: "I thought I was different from everyone and everyone else was better than me. When I looked at that big city, I felt only I'm involved in it. I brought very little and completely cut off relations with everyone. I didn't want to talk to anyone and answer other people's random questions. I didn't want to explain to anyone. I felt like everyone thought it was my fault and. I did. I didn't like others to comment on our situation and give reasons why it happened like this! I didn't want anyone to know anything about his illness because I don't like sympathy at all, to remind my child in the future."
Mother 3 talked about her dependence on cyberspace and her obsessive behavior: "I always had my phone in my hand, I couldn't sleep at all at night, and I kept turning around on the phone. When I put the phone down, I got stressed. I became obsessive. I was sensitive to cleanliness. When I get nervous, I become more obsessive, I work more, and it relaxes me. I was more in control. I felt that others were bothered. I tried to clean myself to relieve myself. I should do something and maintain the conditions; for example, don't say "hey, don't spill" and these words. Everyone was nervous about my actions. My wife said why don't you let us live comfortably; you always make restrictions and make it more difficult. "
The second theme was spiritual issues with the sub-themes of complaining and glorifying God, studying religious books, feeling rejected by God, doubting God's justice, hoping God’s mercy, increasing meditation, and feeling guilty. Mothers with children with leukemia who participated in this study felt guilty and rejected by God. While they doubted God's justice because of their child's cancer, they still had hope for God's grace and read religious books and meditated.
Mother 4 expressed her spiritual challenges like this: "After we found out about my daughter's illness, my relationship with God increased. I had to pray, and I read the Quran more. I talked to God. I never expected my daughter to have such an illness. I felt guilty that maybe I had done something bad. Children are so pure and innocent; why should they get sick like this? I couldn't accept it at all."
Mother 2 also says: "I didn't love anyone and kept saying to God, if you loved me and my daughter, this wouldn't have happened to us. What was our sin?! I kept asking why my daughter became like this! My heart was with God, and I was hopeful, but I was angry with God. Of course, when my daughter's condition improved a little, I calmed down and realized that it was God's will. I tried to ask God to make her better every day. I became hopeful although the first days were a big shock."
The third theme expresses psychological problems with the sub-themes of anger, anxiety, fear, depression, sadness, denial, loneliness, hatred of others, insomnia, and introversion. Having a child with cancer has been a big shock for these families, especially the mothers. It has caused many psychological problems and challenges for these mothers and has had many consequences.
Mother 5 talks about the psychological problems that she experienced because of her child's cancer: "I was under stress, I hated everyone, and I thought I was the only one who got this disease. I sometimes felt nauseous due to extreme sadness and grief. I didn't like to hear anyone's voice or laughter, even in the street. I was depressed and confused. I was constantly depressed and indecisive. Sometimes, from extreme stress and depression, I would stay awake until morning and could not sleep."
Mother 4 also says: "When they diagnosed the disease as definitive, I was very nervous. I was also irritable. In the beginning, I was much stressed, I screamed to control my anger. I was very disappointed; he was my child, and I was afraid of losing him. I used to shake his hand. I was afraid that he wouldn't get better or that the disease would recur and come back. I'm still afraid, I think that this anxiety will be with me for the rest of my life. I used to shout and fight with my husband all the time. I was tired of everything. I didn't have any motivation."
The sub-themes related to the fourth theme, i.e., the challenges related to treatment, included lack of knowledge about the disease, fear of obtaining more information, difficulty in preparing medicine, satisfaction with education and counseling, trust in treatment personnel, satisfaction with the referral system, and respect for privacy. Many of these mothers knew little about their child's illness, but they were afraid to learn more about it. Also, even though they had many problems in preparing medicines, they were satisfied with the referral system, the cooperation of the personnel, and the training and counseling sessions.
Mother 6 says: "I was really shocked when I heard what kind of disease my daughter had. I didn't know that children could get this disease, too! When I was in the hospital, I asked my roommates, but I still couldn't believe it. I thought it was a mistake and that our child didn't have cancer. However, I didn't want to know what would happen next because I was afraid that I would hear all the bad news and lose hope. I wanted to have a positive mindset and did not think about cancer."
Mother 7 talked about the problems of getting medicine: "At the beginning of the illness, we didn't know how to get medicine; it was very difficult, and we faced many problems. Finding good foreign medicine, for example German medicine, was the hardest thing in the world. Besides being very expensive, it was not available at all. We prepared as much as we could; if not, we used any medicine that was available and approved by his doctor."
Mother 8 talked about the cooperation and empathy of the hospital staff and the training and counseling she received: "The thing that gave me hope in the hospital was the treatment staff. Although I think their work was difficult because they had to work with children, they really worked with love and willingness. I was calmed down by their calmness. When we were transferred to a new place, I was worried, but very soon they accepted us and taught us what to do or not to do. They told us what we should do after these medicines were finished."
The sub-themes of increasing economic burden, interruption and closure of the father's work and seeking help from charity foundations also showed the fifth theme, which is economic tensions. Providing medicines and the treatment process has been challenging for families both in terms of cost and time spent, to the extent that, according to many mothers participating in the study, these issues have led to loss or interruptions in the father's work, and they had to seek help from charity foundations for the cost of medicine and treatment.
Mother 9 talked about the economic burden of her husband's illness and unemployment like this: "When we were in the hospital, my husband couldn't go to work properly; I also have another small child and I had to stay in the hospital overnight with my son. Because his immune system was weak, no one could stay with him except his parents. For a few months, we had no income at all because my husband was in trouble. We spent all our savings. After that, when we got under the coverage of the foundation, it was much better. The cost of medication and treatment became much less."
As to the treatment costs, mother 10 said: "Believe me, we were under a lot of pressure economically. We sold our land and car. The drugs were really expensive. We bought 8 drugs for 32 million Tomans. Of course, sometimes we also got drugs through the charity medical center. We realized that we should have shared the cost with someone else."
Discussions
The results of this study showed that the lived experiences of mothers with children with leukemia fell into the five main themes of behavioral problems, spiritual problems, psychological problems, challenges related to treatment, and economic tensions. In this regard, other studies conducted in the English language databases were reviewed, and similar studies were used in this field.
Chronic diseases of children such as leukemia have many negative effects on the lives of children and parents, especially mothers. The child's suffering has a great impact on the parents; they undergo many changes to live with a sick child. In this regard, mothers have to make many adjustments in their lifestyle with a child with leukemia. They have to limit the entry of relatives to the house to prevent the child from getting an infection. Many parents also end up cutting off their relationship with relatives [ 21 ]. In the present study, one of the sub-themes of behavioral problems was disconnection with others, which was mentioned by many participants.
The results of the study by Usha Chivukula and her colleagues (2018) show that there is no significant difference in the burden borne by the parents due to the diagnosis of the disease of their child. However, mothers and fathers use different coping strategies to overcome this crisis and differ in terms of spirituality [ 22 ]. Some mothers feel more optimistic and hopeful about their child's improvement [ 21 ]. Spirituality is an effective strategy for improving the quality of life, psycho-social adaptation to cancer treatment, maintenance of better relationships between caregivers and patients, which indirectly reduces distress and lowers the possibility of poor mental performance in caregivers. It also allows the caregivers to feel positive about their caring role. One of the major correlates for positive emotional state among caregivers of patients with dementia and cancer is the support received from caregivers' religious faith [ 22 ]. In the present study, one of the main themes of the lived experience of mothers with a child with leukemia is spiritual issues. Families who have a very strong faith in God find their faith very supportive. Families feel that their beliefs help them find meaning in their lives, and also give them a sense of confidence that their child will be fine [ 21 ]. Spirituality moderates the adverse effects of stress, and incorporating a spiritual component into coping styles may help develop effective coping interventions. The ability to develop and implement effective coping strategies (tailored to individual needs) is of great importance during a crisis, as it reduces the caregiver’s burden. A multidimensional construct such as spirituality can serve as a reliable resource for coping and improving the caregivers' overall well-being. Identifying the existence of spiritual factors and coping styles and their role in enhancing the quality of life among caregivers is crucial for developing effective coping strategies to match individual needs [ 22 ].
It is said that the physical, mental, and emotional condition of mothers, as one of the main providers of service and care, is affected by the child's illness and creates a lot of stress for them [ 23 ]. Diagnosing and treating cancer in a child causes mental stress, which often has a negative effect on the health of the parents. Studies show that the level of stress and depression, especially anxiety, among parents of children with leukemia is significant [ 24 ]. In line with the present study, anxiety, depression, sadness, and anger are among the sub-themes of psychological problems. In the study of Dana Bakula and her colleagues (2019), it was also reported that parents and children with cancer are at risk of psychological distress, including depression, anxiety and post-traumatic stress [ 8 ]. Usually, mothers are shocked when they hear the diagnosis of their child's disease and go through the process of grief to gradually reach acceptance. Also, many mothers feel emotionally unstable and cry when they hear this diagnosis [ 21 ]. In addition, having a child with cancer is an exhausting lived experience for both parents and causes distress and critical reactions such as shock, disbelief, despair, sadness, and anger [ 25 ]. Also, in this regard, the study of Rezaei et al., which was conducted with the aim of investigating the quality of life of mothers with children with cancer in Iran in 2017, shows that in terms of the components of mental and physical suffering, the condition of mothers with children with cancer is unfavorable [ 23 ]. The findings of another study, which examined the level of stress, anxiety and depression of parents of children with leukemia in a short report, showed that the level of stress and depression, especially anxiety, among parents of children with leukemia was significant [ 24 ].
Many parents have a strong desire to play informational roles and believe that they are in the best position to talk to their child about their illness. Health care professionals have a supportive role to ease the burden of parents who feel responsible for communicating information to their child and other family members. Young children, in particular, rely on their parents for all their medical and non-medical information. Therefore, parents have a heavy responsibility in understanding information, evaluating the appropriate amount of information that should be shared with their child, and then disclosing it. Therefore, parents should first recognize their lack of knowledge [ 26 ]. In the current study, the lack of knowledge about the disease is reported as one of the sub-themes of challenges related to treatment. Although many participants in this study reported fear of learning more about the disease as another sub-theme, a different study found that parents often wanted to learn more even when the information was uncomfortable. Some parents even chose to actively seek information because it helped reduce their uncertainty and increase their sense of control [ 26 ].
Diagnosing cancer in childhood can significantly affect the physical, psycho-social, and socioeconomic well-being of patients and their families. After the first year of diagnosis, having five or more unexpected hospitalizations, that is, unplanned admissions for chemotherapy, leads to greater financial stress for these families. Approximately 20% of families reported more than five unexpected admissions in the first year. Although a specific reason for these unexpected hospitalizations is not known, complications of cancer treatment, such as infection, fever, or septicemia, often increase the need for hospitalization [ 16 ]. Consistent with other studies, the sub-themes of economic tension in the present study include the increase in economic burden, disruption and loss of the father's job, and the need to seek help from charity foundations. Previous research has suggested that treatment-related issues can negatively affect the quality of life and cause long-term emotional, social, and financial stress for parents [ 27 ]. Cancer treatment requires frequent and regular visits of patients as inpatients and outpatients, which often interferes with the work schedule of parents. The results are consistent with the finding that some caregivers have quit or changed careers as a consequence of their child's cancer diagnosis [ 16 ]. Moreover, the treatment of children with cancer is more costly than that of adolescents or adults [ 18 ]. Additionally, existing research indicates that families incur significant variable costs during cancer treatment [ 13 ]. A 2017 Canadian study provides estimates of the economic burden of care during the 90 days before diagnosis and the first year after diagnosis for population-based cohorts of children and adolescents with cancer. The main findings of this study can be summarized in three key points. Firstly, the costs are high in children and adolescents with cancer compared to non-cancer populations. The second thing is that the costs are higher for children than teenagers. Third, the costs for children and teenagers with cancer are higher than adults. Accurate estimation of childhood and adolescent cancer costs provides a valuable scientific basis for cost-effectiveness analyses of cancer treatments in these patients. Cancer care in children and adolescents may be cost-effective despite high costs because the significant effects of cancer treatment on their survival cannot be ignored [ 18 ].
The limitations of the study included the lack of cooperation of some participants in conducting interviews. To address this problem, the researchers tried to establish a good rapport with the mothers.
It is recommended based on the results that mothers should receive accurate information, coping strategies, and motivation to continue treatment through phone counseling. Also, the relevant organizations should be notified of the economic problems of the family, so that appropriate actions can be taken in this area.
Since mothers are regarded as the most important people in the child's support system, children are very sensitive to their behavioral, mental, and emotional state and often mimic their behavior in stressful situations as a way of coping. Based on this study, these mothers experience significant burdens such as behavioral, spiritual, and psychological problems, and financial burden related to treatment and follow-up. The role of parents, especially mothers, as the primary and main caregivers, is crucial in comforting the sick child and taking care of him/her because parental anxiety after cancer diagnosis is one of the factors that lowers the quality of life in a sick child. Therefore, understanding the experiences of parents, especially mothers, in managing and planning for the care of these children seems essential.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Data availability
No datasets were generated or analysed during the current study.
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–49.
Article PubMed Google Scholar
World Health Organization. Cancer: key facts. Fact Sheets. 2018. 2019.
World Health Organization. Childhood cancer: Key facts. 2021.
Steliarova-Foucher E, Colombet M, Ries LA, Moreno F, Dolya A, Bray F, et al. International incidence of childhood cancer, 2001–10: a population-based registry study. Lancet Oncol. 2017;18(6):719–31.
Article PubMed PubMed Central Google Scholar
World Health Organization. CureAll framework: WHO global initiative for childhood cancer: increasing access, advancing quality, saving lives. 2021.
Armstrong GT, Kawashima T, Leisenring W, Stratton K, Stovall M, Hudson MM, et al. Aging and risk of severe, disabling, life-threatening, and fatal events in the childhood cancer survivor study. J Clin Oncol. 2014;32(12):1218.
Smith MA, Seibel NL, Altekruse SF, Ries LA, Melbert DL, O’Leary M, et al. Outcomes for children and adolescents with cancer: challenges for the twenty-first century. J Clin Oncol. 2010;28(15):2625.
Bakula DM, Sharkey CM, Perez MN, Espeleta HC, Gamwell KL, Baudino M, et al. Featured article: The relationship between parent and child distress in pediatric cancer: A meta-analysis. J Pediatr Psychol. 2019;44(10):1121–36.
Hu X, Grosse SD, Han X, Marchak JG, Ji X. Mental Health Care Utilization Among Parents of Children With Cancer. JAMA Netw Open. 2024;7(4):e244531-e.
Article Google Scholar
Allen R, Newman S, Souhami R. Anxiety and depression in adolescent cancer: findings in patients and parents at the time of diagnosis. Eur J Cancer. 1997;33(8):1250–5.
Article CAS PubMed Google Scholar
Boman KK, Viksten J, Kogner P, Samuelsson U. Serious illness in childhood: the different threats of cancer and diabetes from a parent perspective. J Pediatr. 2004;145(3):373–9.
Katz LF, Fladeboe K, King K, Gurtovenko K, Kawamura J, Friedman D, et al. Trajectories of child and caregiver psychological adjustment in families of children with cancer. Health Psychol. 2018;37(8):725.
Tsimicalis A, Stevens B, Ungar WJ, McKeever P, Greenberg M. The cost of childhood cancer from the family’s perspective: a critical review. Pediatr Blood Cancer. 2011;56(5):707–17.
Atun R, Bhakta N, Denburg A, Frazier AL, Friedrich P, Gupta S, et al. Sustainable care for children with cancer: a Lancet Oncology Commission. Lancet Oncol. 2020;21(4):e185–224.
Essue BM, Iragorri N, Fitzgerald N, de Oliveira C. The psychosocial cost burden of cancer: A systematic literature review. Psychooncology. 2020;29(11):1746–60.
Warner EL, Kirchhoff AC, Nam GE, Fluchel M. Financial burden of pediatric cancer for patients and their families. Journal of oncology practice. 2015;11(1):12–8.
Wiemels J. Perspectives on the causes of childhood leukemia. Chem Biol Interact. 2012;196(3):59–67.
De Oliveira C, Bremner KE, Liu N, Greenberg ML, Nathan PC, McBride ML, Krahn MD. Costs for childhood and adolescent cancer, 90 days prediagnosis and 1 year postdiagnosis: a population-based study in Ontario. Canada Value in health. 2017;20(3):345–56.
Larkin M, Flowers P, Smith JA. Interpretative phenomenological analysis: Theory, method and research. Interpretative phenomenological analysis. 2021:1–100.
Schwandt TA, Lincoln YS, Guba EG. Judging interpretations: But is it rigorous? Trustworthiness and authenticity in naturalistic evaluation. N Dir Eval. 2007;2007(114):11–25.
Cornelio SJ, Nayak BS, George A. Experiences of mothers on parenting children with leukemia. Indian J Palliat Care. 2016;22(2):168.
Chivukula U, Kota S, Nandinee D. Burden experience of caregivers of acute lymphoblastic leukemia: Impact of coping and spirituality. Indian J Palliat Care. 2018;24(2):189.
Rezaei Z, Sani MS, Ostadhashemi L, Harouni GG. Quality of life of mothers of children with cancer in Iran. Koomesh. 2018;20(3):425–31.
Google Scholar
Farhangi H, Mohareri F, Jarahi L, Armanpoor P. Evaluation of stress, anxiety and depression in parents of children with leukemia: Brief report. Tehran Univ Med Sci J. 2017;74(10):741–5.
Kars MC, Duijnstee MS, Pool A, Van Delden JJ, Grypdonck MH. Being there: parenting the child with acute lymphoblastic leukaemia. J Clin Nurs. 2008;17(12):1553–62.
Gibson F, Kumpunen S, Bryan G, Forbat L. Insights from parents of a child with leukaemia and healthcare professionals about sharing illness and treatment information: A qualitative research study. Int J Nurs Stud. 2018;83:91–102.
Seth L. Lived Experiences of Support Among Parents of Children With Acute Lymphoblastic Leukemia During the COVID-19 Pandemic: Walden University (Doctoral dissertation); 2022.
Download references
Acknowledgements
The authors would like to thank all participants who willingly participated in this study.
This research was performed with the financial support of Shiraz University of Medical Sciences (SUMS), Shiraz, Iran.
Author information
Authors and affiliations.
Student Research Committee, Shiraz University of Medical Sciences, Shiraz, Iran
Fatemeh Shaygani
Health Policy Research Center, Institute of Health, Shiraz University of Medical Sciences, Shiraz, Iran
Fatemeh Shaygani & Hana Javanmardi Fard
Department of Medical Education, Clinical Education Research Center, School of Medicine, Shiraz University of Medical Sciences, Shiraz, Iran
Katayoun Jalali
Midwifery Department, Estahban Branch, Islamic Azad University, Estahban, Iran
Zahra Afrasiabi
Department of Health in Disasters and Emergencies, Health Human Resources Research Center, School of Health Management and Information Sciences, Shiraz University of Medical Sciences, Shiraz, Iran
Milad Ahmadi Marzaleh
You can also search for this author in PubMed Google Scholar
Contributions
F.Sh. conducted the literature research for the background of the study, planned the study, analyzed and interpreted data, and contributed to the writing of the article. K.J planned the study, analyzed, and interpreted data, and wrote the article. H.J.F. and Z.A. collected data, and contributed analyzed and interpreted data. M.A.M.supervised the study, revised the article and proofread the manuscript. The authors read and approved the final manuscript.
Corresponding author
Correspondence to Katayoun Jalali .
Ethics declarations
Ethics approval and consent to participate.
The ethical approval for the study was acquired from the Research Ethics Committee of Shiraz University of Medical Sciences (IR.SUMS.NUMIMG.REC.1402.051). All methods were carried out in accordance with relevant guidelines and regulations or the Declaration of Helsinki. Participants were given detailed information about the study’s goals before interviews were conducted, and their signed informed consent was obtained before any interviews were recorded. The anonymity of the participants, their responses, and the use of aliases or codes in quotations were guaranteed. Participation in the research was entirely voluntary and they were given the option to drop out at any time. Both the interview and encoding data were encrypted before being saved to a personal hard drive for long-term storage. We promised to do so if a participant wanted to see the findings as a group.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note.
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/ .
Reprints and permissions
About this article
Cite this article.
Shaygani, F., Jalali, K., Javanmardi Fard, H. et al. Exploring the lived experience of mothers of children with leukemia: a qualitative study from Iran. BMC Women's Health 24 , 457 (2024). https://doi.org/10.1186/s12905-024-03300-y
Download citation
Received : 20 November 2023
Accepted : 08 August 2024
Published : 16 August 2024
DOI : https://doi.org/10.1186/s12905-024-03300-y
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
- Lived experience
- Pediatric cancer
BMC Women's Health
ISSN: 1472-6874
- Submission enquiries: [email protected]
- General enquiries: [email protected]
IMAGES
COMMENTS
Case study submitted by Hanny Al-Samkari, MD, of Massachusetts General Hospital, Dana-Farber Cancer Institute, Harvard University, Boston, MA References Stone RM, Mandrekar SJ, Sanford BL, et al. Midostaurin plus Chemotherapy for Acute Myeloid Leukemia with a FLT3 Mutation .
Rare and emerging subtypes of leukemia can be incredibly challenging to diagnose and even more challenging to treat. At the NCCN 2019 Annual Congress: Hematologic Malignancies, a panel of experts, moderated by Andrew D. Zelenetz, MD, PhD, were presented with particularly challenging cases in these malignancies and asked to discuss best approaches to treatment.
A 49-year-old man was evaluated because of relapsed acute myeloid leukemia that occurred 14 months after the initial diagnosis and treatment. Molecular genetic profiling at the time of relapse iden...
Adolescents and young adults (AYA) diagnosed with acute lymphoblastic leukemia (ALL) have faced poorer survival rates compared with the history of this illness treatment in children [].However, several European and US studies have reported improved outcomes for AYA patients treated with pediatric-based protocols [2,3,4]. however, AYA patients receiving pediatric regimens and doses, unlike ...
Acute myeloid leukemia developed in a woman approximately 5.5 years after she had received LentiGlobin for sickle cell disease as part of the initial cohort (Group A) of the HGB-206 study.
Chronic lymphocytic leukemia (CLL) is one of the most common types of leukemia among adults in the United States and is still considered incurable. 1,2 It affects B and T lymphocytes as well as natural killer cells, but the majority of CLL cases diagnosed are of the B-cell phenotype. 3 CLL results from the uncontrolled clonal growth of small B lymphocytes in a manner that often leads to the ...
In this case, we can confirm an increase in mature WBCs, with a predominance of neutrophils and their precursors. Compare this to a case of acute leukemia (Figure 2), which shows a predominance of immature cells, or blasts, characterized by increased nuclear:cytoplasmic ratios, immature chromatin, nucleoli, and lack of nuclear segmentation ...
Introduction. Philadelphia chromosome-positive acute lymphoblastic leukemia (Ph+ ALL) is defined by the translocation t(9;22)(q34; q11,2) and accounts for 2%-5% of pediatric and 25%-30% of adult ALL cases ().Of note, Ph+ ALL is almost exclusively of B lineage, with less than 2% of cases of T lineage in the pediatric Children Oncology Group (COG) and in the Ph+ ALL European group (EsPhALL ...
Case Study: New Therapies for Acute Myeloid Leukemia. A 76-year-old woman presents to the emergency department following two weeks of progressive dyspnea and fatigue, and a new rash. Her medical history is significant for stage 2 chronic kidney disease, coronary artery disease, and diabetes. Physical examination results are within normal limits ...
Background Acute lymphoblastic leukemia is the most common type of cancer in children. Most often it affects the age group between 2 and 5 years of age. Studies have shown an improvement in general survivability, more than 90% 5-year overall survival (OS). Current treatment protocols for acute lymphoblastic leukemia require verification of the presence of favorable and unfavorable genetic ...
s is an open accessCase PresentationA 67-year-old woman with a past medical history of hypertension, valvular heart disease, and dyslipidemia presented with fatigue, dysp. ea, decreased appetite, and jaundice. There was laboratory evidence of. emolytic anemia and thrombocytopenia. A bone marrow biopsy was consistent with acute myeloid leukem.
This interactive case report will engage your experience and knowledge about the details of a given case. Along the way, we will provide details and at the end, we will share the patient's outcome. Acute Myeloid Leukemia Infiltration of the Stomach (Consultant. 2017;57 [1]:54-55) A 41-year-old man with a past medical history of hypertension ...
Background Blast transformation is a rare but well-recognized event in Philadelphia-negative myeloproliferative neoplasms associated with a poor prognosis. Secondary acute myeloid leukemias evolving from myeloproliferative neoplasms are characterized by a unique set of cytogenetic and molecular features distinct from de novo disease. t(8;21) (q22;q22.1); RUNX1::RUNX1T1, one of the most ...
1 Introduction. Acute myeloid leukemia (AML) is a malignantly clonal disorder characterized by blockage of differentiation in the myeloid lineage and an accumulation of its immature progenitors in bone marrow, leading to hematopoietic failure. In China, it was predicted that there were about 75,300 newly diagnosed leukemia cases in 2015; meanwhile, it was estimated that about 53,400 Chinese ...
AML-M6 appears to be bilineage leukemia; our case initially presented as pure erythroleukemia with 80-90% erythroblasts and less than 5% myeloblasts, but after treatment with chemotherapy, it transformed into AML with an increase in myeloblasts up to 30%. ... a Cancer and Leukemia Group B Study. J Clin Oncol. 2007;25:3337-3343. 17. Schlenk RF ...
834 n engl j med 385;9 nejm.org August 26, 2021 Te e nglan ourna o edicine Presentation of Case Dr. Richard A. Newcomb: A 49-year-old man was evaluated at this hospital because of relapsed acute ...
Background. PDGFRB fusions in acute lymphoblastic leukemia (ALL) is rare. The authors identified 28 pediatric PDGFRB-positive ALL.They analyzed the features, outcomes, and prognostic factors of this disease. Methods. This multicenter, retrospective study included 6457 pediatric patients with newly diagnosed PDGFRB fusion ALL according to the CCCG-ALL-2015 and CCCG-ALL-2020 protocols from April ...
In contrast to the combination approach taken by MDACC and EWALL, the German Multicenter Study Group for Adult Acute Lymphoblastic Leukemia (GMALL) study group is studying a sequential approach. In the phase 2 INITIAL-1 study (NCT03460522), untreated older adults (aged >55 years) receive 3 cycles of IO monotherapy followed by CC consolidation ...
Initial Presentation. A 67-year-old man presented to PCP with complaints of fatigue and night sweats. PMH: patient takes OTC antiacid tablets a few times a week for a "sensitive" stomach. PE: Enlarged mobile lymph nodes bilaterally (~1.5 cm), no palpable spleen or liver. Laboratory findings: WBC; 102 X 109/L.
Abstract. We report two cases of chronic myeloid leukaemia (CML) with extreme thrombocytosis. The first patient was a 65-year-old man who presented with prolonged history of upper abdominal discomfort, anorexia and two episodes of recent gum bleeds without fever or other bleeding manifestations. He was a chronic smoker with no other comorbidities.
Case Study: Prognostic Factors in Acute Lymphocytic Leukemia. A 48-year-old female presented to the emergency department with severe headaches, dyspnea on exertion, and petechiae on the lower extremities. A CBC was drawn that showed the following: WBC=56 x10 3 /µL, Hgb=9.0 g/dL, Hct=23, MCV=97 fl, plt=15 x10 9 /µL, ANC=0.7x10 3 /µL.
In this case, researchers studied more than 1,300 patients treated on the COG AALL0434 clinical trial and sequenced both the tumor and non-tumor genomes of each patient. While the researchers previously suspected that non-coding DNA in T-ALL played an important role, this study's findings are the first ever to establish that at a large scale.
Khalid A, Aslam S, Ahmed M, Hasnain S, Aslam A. Risk assessment of FLT3 and PAX5 variants in B-acute lymphoblastic leukemia: a case-control study in a Pakistani cohort. Peer J. 2019;7:e7195. Crossref. Google Scholar. 11. McGowan-Jordan J, Simons A, Schmid M, eds. ISCN 2016: An International System for Human Cytogenomic Nomenclature (2016). S ...
Sep. 1, 2022 — Scientists have created a roadmap of the genetic mutations present in the most common childhood cancer, acute lymphoblastic leukemia (ALL). The study supplies a comprehensive view ...
December 23, 2019. Video. Naval G. Daver, MD: This is the case of a 64-year-old patient with newly diagnosed acute myeloid leukemia. The patient has significant comorbidities, including an elevated BMI [body mass index], signifying a significant obesity, as well as underlying pneumonia and prior history of high blood pressure and diabetes.
Genomic classification of ETP/ETP-like ALL. Credit: Nature (2024). DOI: 10.1038/s41586-024-07807-
Discussion. The incidence of acute leukemia is approximately 2.3 per 100000 people per year.AML M-7 is a rare subtype of leukemia and represents 1.2% of cases of adult leukemia, compared to 3-10% of childhood leukemia ( 10 ).It is classified under M-7 in the French-American-British classification ( 11 ). The patient was a 26 month years old ...
Acute Lymphocytic Leukemia Case Study. Leukemia is a cancer of white blood cells. In acute leukemia, the abnormal cells divide rapidly, quickly overtaking functional white and red blood cells. The most common form of cancer in children 0-14 years of age is acute lymphocytic leukemia (ALL). The survival rate in children has improved more than 50 ...
Discussion. Acute lymphoblastic leukaemia (ALL) is the most common malignancy in the paediatric age group. 1 Symptoms of ALL include anaemia, fever, bleeding tendency and fatigue. 2 Hypercalcaemia and osteolytic lesions are rare in B-ALL in contrast to their incidence in some other lymphoid malignancies like adult T-cell leukaemia/lymphoma, myeloma and LCH. 2-4 However, in our case, due to ...
Leukemia, as one of the most common pediatric cancers, has negatively affected many children around the world. Parents often experience increased feeling of distress shortly after being informed about their child's diagnosis. The distress experienced by parents can adversely affect various aspects of their life. This study aimed to develop an understanding of the lived experience of the ...