Navigation Menu
Search code, repositories, users, issues, pull requests..., provide feedback.
We read every piece of feedback, and take your input very seriously.

Saved searches
Use saved searches to filter your results more quickly.
To see all available qualifiers, see our documentation .
- Notifications You must be signed in to change notification settings
Application to assign secondary structure to proteins
PDB-REDO/dssp
Folders and files, repository files navigation.
This is a rewrite of DSSP, now offering full mmCIF support. The difference with previous releases of DSSP is that it now writes out an annotated mmCIF file by default, storing the secondary structure information in the _struct_conf category.
Another new feature in this version of DSSP is that it now defines Poly-Proline helices as well.
The DSSP program was designed by Wolfgang Kabsch and Chris Sander to standardize secondary structure assignment. DSSP is a database of secondary structure assignments (and much more) for all protein entries in the Protein Data Bank (PDB). DSSP is also the program that calculates DSSP entries from PDB entries.
DSSP does not predict secondary structure.
Requirements
A good, modern compiler is needed to build the mkdssp program since it uses many new C++20 features.
The new makefile for dssp will take care of downloading and building all requirements automatically. So in theory, building is as simple as:
See manual page for more info. Or even better, see the DSSP website .
Contributors 3
- CMake 14.1%
Protein secondary structure classification revisited: processing DSSP information with PSSC
Affiliation.
- 1 Fachbereich Biologie, Chemie, Pharmazie/Institute of Chemistry and Biochemistry, Freie Universität Berlin , Fabeckstrasse 36A, 14195 Berlin, Germany.
- PMID: 24866861
- DOI: 10.1021/ci5000856
A first step toward three-dimensional protein structure description is the characterization of secondary structure. The most widely used program for secondary structure assignment remains DSSP, introduced in 1983, with currently more than 400 citations per year. DSSP output is in a one-letter representation, where much of the information on DSSP's internal description is lost. Recently it became evident that DSSP overlooks most π-helical structures, which are more prevalent and important than anticipated before. We introduce an alternative concept, representing the internal structure characterization of DSSP as an eight-character string that is human-interpretable and easy to parse by software. We demonstrate how our protein secondary structure characterization (PSSC) code allows for inspection of complicated structural features. It recognizes ten times more π-helical residues than does the standard DSSP. The plausibility of introduced changes in interpreting DSSP information is demonstrated by better clustering of secondary structures in (φ, ψ) dihedral angle space. With a sliding sequence window (SSW), helical assignments with PSSC remain invariant compared with an assignment based on the complete structure. In contrast, assignment with DSSP can be changed by residues in the neighborhood that are in fact not interacting with the residue under consideration. We demonstrate how one can easily define new secondary structure classification schemes with PSSC and perform the classifications. Our approach works without changing the DSSP source code and allows for more detailed protein characterization.
- Computational Biology / methods*
- Models, Molecular
- Protein Structure, Secondary
- Proteins / chemistry*
If you use the DSSP software or databank please cite the appropriate paper:
Using DSSP data
DSSP provides an elaborate description of the secondary structure elements in a protein structure, including backbone hydrogen bonding and the topology of β-sheets. The most popular feature is the per-residue assignment of secondary structure with a single character code:
- H = α-helix
- B = residue in isolated β-bridge
- E = extended strand, participates in β ladder
- G = 3 10 -helix
- I = π-helix
- P = κ-helix (poly-proline II helix)
- T = hydrogen-bonded turn
The full DSSP output is provided in two formats. The legacy DSSP format was origianlly designed for structures that were in PDB-formatted models. Now, 40 years later, the PDB format has become obsolete as it cannot capture the large structure models that modern structural biology methods can provide. The mmCIF format is the data format of choice for structural biology as it has no size limitations for structure models and it can hold extensive annotations and metadata. DSSP now writes its data straight to these mmCIF files by default. The legacy DSSP format can still be written but only for structure models that fit.
DSSP format
The output from DSSP contains secondary structure assignments and other information. Extract from 3kew.dssp (header):
The first few lines are taken from the input model file, then some general statistics about the model and hydrogen bonding are given. The histograms describe the distribution of sizes of secondary structure elements. For instance, this structure has three helices, one short one consisting of 4 residues and two longer ones of 16 and 17 residues. Note that beta sheets are described as a collection of ladders, rather than strands. Ladders can be seen as two strands together with the hydrogen bonds as the rungs of the ladder. More formal definitions are given in the Kabsch and Sander paper.
The model statistics are followed by a detailed per-residue description. Extract from 3kew.dssp (continued):
Below is a brief description of the data columns. More details are described in the Kabsch and Sander paper.
Two columns of residue numbers. First column is DSSP's sequential residue number, starting at the first residue actually in the model set and including chain breaks; this number is used to refer to residues throughout. The second column gives the numbering as is used in the structure model 'residue number','insertion code' and 'chain identifier'; these are given for reference only.
One letter amino acid code, non standard residues are marked as X . CYS in an SS-bridge are marked by a lower case letter. So when cysteines are bridged, then the first bridged cysteine in the sequence and its partner elsewhere in the sequence are marked a . The next bridged cysteine, that is not yet marked, and its partner are both marked b , etcetera. Unbridged cysteines remain marked as C .
The first column (under the S ) gives aone-letter summary of secondary structure, intended to approximate crystallographers' intuition. This summary is based on the next 8 columns, which are the principal result of DSSP analysis of the atomic coordinates. More details in the Kabsch and Sander paper.
BP1 and BP2
Residue numbers of the first and (if available) second beta bridge partner. The letter marked the B-sheet that contains the bridges.
- Effects leading to larger than expected values: solvent exposure calculation ignores unusual residues, like ACE, or residues with incomplete backbone. it also ignores HETATOMS, like a heme or metal ligands. Also, side chains may not have all atoms explicitly modeled.
- Effects leading to smaller than expected values: in complexes, e.g. a dimer, solvent exposure is for the entire assembly, not for the monomer. Also, atom OXT of c-terminal residues is treated like a side chain atom if it is listed as part of the last residue.
- Unknown or non-standard residues are named X on output and are not checked for the expected number of sidechain atoms.
- All explicit water molecules, like other hetatoms, are ignored.
N-H-->O etc.
Hydrogen bonds; e.g. -3,-1.4 means that this residue (i) has its HN atom H-bonded to O of residue i-3 with an electrostatic H-bond energy of -1.4 kcal/mol. There are two columns for each type of H-bond, to allow for bifurcated H-bonds. Note: The marked H-bonds are the best and second best candidate. The second best and even the best (in rare occasions) may be unrealistically por H-bonds.
The cosine of angle between C=O of residue i and C=O of residue i-1. For α-helices, TCO is near +1, for β-sheets TCO is near -1. These values are descriptive and not used for structure definition.
Virtual bond angle (bend angle) defined by the three Cα atoms of residues i-2, i, and i+2. Used to define bends (structure code S ).
Virtual torsion angle (dihedral angle) defined by the four Cα atoms of residues i-1, i, i+1, and i+2. Used to define chirality (structure code + or - ).
PHI and PSI
The peptide backbone torsion angles as described in the IUPAC standard
X-CA, Y-CA, and Z-CA
Just a copy of the Cα atom coordinates in the structure model
DSSP data in mmCIF files
The mmCIF-formatted DSSP output caries the same information as the DSSP format but in a more scalable way and with a formal description caputered in an mmCIF dictionary. It is designed to be machine readable. Developers who create software to read these annotations can use our extension to the mmCIF dictionary on GitHub. Note: For sake of speed the solvent accessibility is not calculated by default when using mmCIF output. The command-line switch --calculate-accessibility can be used to switch this feature on.
Assigning secondary structure in proteins using AI
- Original Paper
- Published: 17 August 2021
- Volume 27 , article number 252 , ( 2021 )
Cite this article

- Jisna Vellara Antony ORCID: orcid.org/0000-0001-5210-9583 1 ,
- Prayagh Madhu 2 ,
- Jayaraj Pottekkattuvalappil Balakrishnan ORCID: orcid.org/0000-0002-9924-9046 1 &
- Hemant Yadav 1
489 Accesses
4 Citations
Explore all metrics
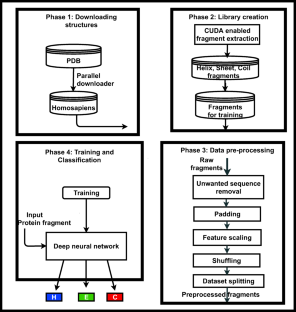
Knowledge about protein structure assignment enriches the structural and functional understanding of proteins. Accurate and reliable structure assignment data is crucial for secondary structure prediction systems. Since the 1980s, various methods based on hydrogen bond analysis and atomic coordinate geometry, followed by machine learning, have been employed in protein structure assignment. However, the assignment process becomes challenging when missing atoms are present in the protein files. Our method proposed a multi-class classifier program named DLFSA for assigning protein secondary structure elements (SSE) using convolutional neural networks (CNNs). A fast and efficient GPU-based parallel procedure extracts fragments from protein files. The model implemented in this work is trained with a subset of the protein fragments and achieves 88.1% and 82.5% train and test accuracy, respectively. The model uses only C α coordinates for secondary structure assignments. The model has been successfully tested on a few full-length proteins also. Results from the fragment-based studies demonstrate the feasibility of applying deep learning solutions for structure assignment problems.
This is a preview of subscription content, log in via an institution to check access.
Access this article
Price includes VAT (Russian Federation)
Instant access to the full article PDF.
Rent this article via DeepDyve
Institutional subscriptions
Similar content being viewed by others

Deep Learning: A Comprehensive Overview on Techniques, Taxonomy, Applications and Research Directions

Review of deep learning: concepts, CNN architectures, challenges, applications, future directions

CBAM: Convolutional Block Attention Module
Data availability.
DLFSA is made available to the public through the Web portal— www.proteinallinfo.in . The datasets generated during and/or analyzed during the current study are not publicly available due to their large size but are available from the corresponding author on reasonable request.
Pauling L, Corey RB, Branson HR (1951) The structure of proteins: two hydrogen-bonded helical configurations of the polypeptide chain. Proc Natl Acad Sci 37(4):205–211
Article CAS PubMed PubMed Central Google Scholar
Reeb J, Rost B (2019) Secondary structure prediction. Encyclopedia of Bioin-formatics and Computational Biology, pp 488–496
Chapter Google Scholar
Srinivasan R, Rose GD (1999) A physical basis for protein secondary structure. Proc Natl Acad Sci 96(25):14258–14263
Eisenberg D (2003) The discovery of the α-helix and β-sheet, the principal structural features of proteins. Proc Natl Acad Sci 100(20):11207–11210
Zhou J, Wang H, Zhao Z, Xu R, Lu Q (2018) CNNH_PSS: protein 8-class secondary structure prediction by convolutional neural network with highway. BMC Bioinform 19(4):99–109
Google Scholar
Abbass J, Nebel JC, Mansour N, Elloumi M, Zomaya AY (2013) Ab initio protein structure prediction: methods and challenges. Biol Knowl Discov Handb. John Wiley & Sons, Inc, Hoboken, New Jersey, pp 703–724
Anfinsen CB (1973) Principles that govern the folding of protein chains. Science 181(4096):223–230
Article CAS PubMed Google Scholar
Onuchic JN, Wolynes PG (2004) Theory of protein folding. Curr Opin Struct Biol 14(1):70–75
Kabsch W, Sander C (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22(12):2577–2637
Frishman D, Argos P (1995) Knowledge-based protein secondary structure assignment. Proteins Struct Funct Bioinf 23(4):566–579
Article CAS Google Scholar
Ramachandran GT, Sasisekharan V (1968) Conformation of polypeptides and proteins. Adv Protein Chem 23:283–437
Zacharias J, Knapp EW (2014) Protein secondary structure classification revisited: processing DSSP information with PSSC. J Chem Inf Model 54(7):2166–2179
Fodje MN, Al-Karadaghi S (2002) Occurrence, conformational features and amino acid propensities for the π-helix. Protein Eng Des Sel 15(5):353–358
Nagy G, Oostenbrink C (2014) Dihedral-based segment identification and classification of biopolymers I: proteins. J Chem Inf Model 54(1):266–277
Cubellis MV, Cailliez F, Lovell SC (2005) Secondary structure assignment that accurately reflects physical and evolutionary characteristics. BMC Bioinform 6(4):1–9
Richards FM, Kundrot CE (1988) Identification of structural motifs from protein coordinate data: secondary structure and first-level supersecondary structure. Proteins Struct Funct Bioinf 3(2):71–84
Sklenar H, Etchebest C, Lavery R (1989) Describing protein structure: a general algorithm yielding complete helicoidal parameters and a unique overall axis. Proteins Struct Funct Bioinf 6(1):46–60
Hosseini SR, Sadeghi M, Pezeshk H, Eslahchi C, Habibi M (2008) PROSIGN: a method for protein secondary structure assignment based on three-dimensional coordinates of consecutive Cα atoms. Comput Biol Chem 32(6):406–411
Labesse G, Colloc'h N, Pothier J, Mornon JP (1997) P-SEA: a new efficient assignment of secondary structure from Cα trace of proteins. Bioinformatics 13(3):291–295
Majumdar I, Krishna SS, Grishin NV (2005) PALSSE: a program to delineate linear secondary structural elements from protein structures. BMC Bioinform 6(1):1–24
Taylor WR (2001) Defining linear segments in protein structure. J Mol Biol 310(5):1135–1150
Dupuis F, Sadoc JF, Mornon JP (2004) Protein secondary structure assignment through Voronoi tessellation. Proteins Struct Funct Bioinf 55(3):519–528
Park SY, Yoo MJ, Shin JM, Cho KH (2011) SABA (secondary structure assignment program based on only alpha carbons): a novel pseudo center geometrical criterion for accurate assignment of protein secondary structures. BMB Rep 44(2):118–122
Zhang W, Dunker AK, Zhou Y (2008) Assessing secondary structure assignment of protein structures by using pairwise sequence-alignment benchmarks. Proteins Struct Funct Bioinf 71(1):61–67
Cao C, Wang G, Liu A, Xu S, Wang L, Zou S (2016) A new secondary structure assignment algorithm using Cα backbone fragments. Int J Mol Sci 17(3):333
Article PubMed PubMed Central CAS Google Scholar
Konagurthu AS, Lesk AM, Allison L (2012) Minimum message length inference of secondary structure from protein coordinate data. Bioinformatics 28(12):i97–i105
Haghighi H, Higham J, Henchman RH (2016) Parameter-free hydrogen-bond definition to classify protein secondary structure. J Phys Chem B 120(33):8566–8570
Kumar P, Bansal M (2012) HELANAL-Plus: a web server for analysis of helix geometry in protein structures. J Biomol Struct Dyn 30(6):773–783
King SM, Johnson WC (1999) Assigning secondary structure from protein coordinate data. Proteins Struct Funct Bioinf 35(3):313–320
Carter P, Andersen CA, Rost B (2003) DSSPcont: continuous secondary structure assignments for proteins. Nucleic Acids Res 31(13):3293–3295
Konagurthu AS, Allison L, Stuckey PJ, Lesk AM (2011) Piecewise linear approximation of protein structures using the principle of minimum message length. Bioinformatics 27(13):i43–i51
Levitt M, Greer J (1977) Automatic identification of secondary structure in globular proteins. J Mol Biol 114(2):181–239
Cao C, Xu S, Wang L (2015) An algorithm for protein helix assignment using helix geometry. PLoS One 10(7):e0129674
Klose DP, Wallace BA, Janes RW (2010) 2Struc: the secondary structure server. Bioinformatics 26(20):2624–2625
Kumar P, Bansal M (2015) Identification of local variations within secondary structures of proteins. Acta Crystallogr D Biol Crystallogr 71(5):1077–1086
Habibia M, Eslahchia C, Pezeshkc H, Sadeghid M (2008) An information-theoretic approach to secondary structure assignment, Journal of Science (University of Tehran) (JSUT)
Taylor T, Rivera M, Wilson G, Vaisman II (2005) New method for protein secondary structure assignment based on a simple topological descriptor. Proteins Struct Funct Bioinf 60(3):513–524
Zhang Y, Sagui C (2015) Secondary structure assignment for conformationally irregular peptides: comparison between DSSP, STRIDE and KAKSI. J Mol Graph Model 55:72–84
Article PubMed CAS Google Scholar
Law SM, Frank AT, Brooks III CL (2014) PCASSO: a fast and efficient Cα-based method for accurately assigning protein secondary structure elements. J Comput Chem 35(24):1757–1761
Salawu EO (2016) RaFoSA: Random forests secondary structure assignment for coarse-grained and all-atom protein systems. Cogent Biol 2(1):1214061
Wang J, Cao H, Zhang JZ, Qi Y (2018) Computational protein design with deep learning neural networks. Sci Rep 8(1):1–9
Cheng J, Tegge AN, Baldi P (2008) Machine learning methods for protein structure prediction. IEEE Rev Biomed Eng 1:41–49
Article PubMed Google Scholar
Zhang B, Li J, Lü Q (2018) Prediction of 8-state protein secondary structures by a novel deep learning architecture. BMC Bioinform 19(1):1–13
LeCun Y, Bengio Y, Hinton G (2015) Deep learning. Nature 521(7553):436–444
Goh GB, Hodas NO, Vishnu A (2017) Deep learning for computational chemistry. J Comput Chem 38(16):1291–1307
O'Shea, K., & Nash, R. (2015). An introduction to convolutional neural networks. arXiv preprint arXiv:1511.08458.
Busia, A., Collins, J., & Jaitly, N. (2016). Protein secondary structure prediction using deep multi-scale convolutional neural networks and next-step conditioning. arXiv preprint arXiv:1611.01503.
Zamora-Resendiz R, Crivelli S (2019) Structural learning of proteins using graph convolutional neural networks. bioRxiv, 610444, Cold Spring Harbor Laboratory
Niepert, M., Ahmed, M., & Kutzkov, K. (2016). Learning convolutional neural networks for graphs. In International conference on machine learning (pp. 2014-2023). PMLR.
https://www.rcsb.org/structure/ , accessed : 2020-09-09.
Holmes JB, Tsai J (2004) Some fundamental aspects of building protein structures from fragment libraries. Protein Sci 13(6):1636–1650
Xu D, Zhang Y (2013) Toward optimal fragment generations for ab initio protein structure assembly. Proteins Struct Funct Bioinf 81(2):229–239
de Oliveira SH, Shi J, Deane CM (2015) Building a better fragment library for de novo protein structure prediction. PLoS One 10(4):e0123998
Abbass J, Nebel JC (2015) Customised fragments libraries for protein structure prediction based on structural class annotations. BMC Bioinform 16(1):1–13
Trevizani R, Custódio FL, Dos Santos KB, Dardenne LE (2017) Critical features of fragment libraries for protein structure prediction. PLoS One 12(1):e0170131
Abbass J, Nebel JC (2020) Enhancing fragment-based protein structure prediction by customising fragment cardinality according to local secondary structure. BMC Bioinform 21:1–23
https://www.djangoproject.com/ , accessed : 2020-12-12.
Download references
Acknowledgements
The authors would like to thank the Central Computing Centre, National Institute of Technology Calicut (NITC), for providing GPU servers for this work. The authors would like to acknowledge the valuable suggestions from Jinto Antony for developing the Web portal.
Code availability
The library generation source codes and the model codes are made open at https://github.com/jisnava/DLFSA/ .
It is part of my (VA Jisna) PhD work at the National Institute of Technology Calicut, India. The research is funded by the Ministry of Human Resource Development, India.
Author information
Authors and affiliations.
Department of Computer Science and Engineering, National Institute of Technology Calicut, Kerala, 673601, India
Jisna Vellara Antony, Jayaraj Pottekkattuvalappil Balakrishnan & Hemant Yadav
Computer Science and Engineering Dept., Rajiv Gandhi Institute of Technology, Kottayam, India
Prayagh Madhu
You can also search for this author in PubMed Google Scholar
Contributions
Jisna Vellara Antony (JVA) did the conceptualization. Hemant Yadav developed the fragment library. Prayagh Madhu did the model coding. JVA developed the Web portal and wrote the manuscript. Jayaraj Pottekkattuvalappil Balakrishnan supervised the project. All authors read and approved the final manuscript.
Corresponding author
Correspondence to Jisna Vellara Antony .
Ethics declarations
Competing interests.
The authors declare no competing interests.
Additional information
Publisher’s note.
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
(DOCX 335 kb)
Rights and permissions
Reprints and permissions
About this article
Antony, J.V., Madhu, P., Balakrishnan, J.P. et al. Assigning secondary structure in proteins using AI. J Mol Model 27 , 252 (2021). https://doi.org/10.1007/s00894-021-04825-x
Download citation
Received : 22 January 2021
Accepted : 16 June 2021
Published : 17 August 2021
DOI : https://doi.org/10.1007/s00894-021-04825-x
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
- Protein structure assignment
- Deep learning
- Fragment library creation
- Convolutional neural networks
- Protein fragments
- Protein secondary structures
- Multi-class classifier
- Find a journal
- Publish with us
- Track your research
- Search Menu
- Sign in through your institution
- Chemical Biology and Nucleic Acid Chemistry
- Computational Biology
- Critical Reviews and Perspectives
- Data Resources and Analyses
- Gene Regulation, Chromatin and Epigenetics
- Genome Integrity, Repair and Replication
- Methods Online
- Molecular Biology
- Nucleic Acid Enzymes
- RNA and RNA-protein complexes
- Structural Biology
- Synthetic Biology and Bioengineering
- Advance Articles
- Breakthrough Articles
- Special Collections
- Scope and Criteria for Consideration
- Author Guidelines
- Data Deposition Policy
- Database Issue Guidelines
- Web Server Issue Guidelines
- Submission Site
- About Nucleic Acids Research
- Editors & Editorial Board
- Information of Referees
- Self-Archiving Policy
- Dispatch Dates
- Advertising and Corporate Services
- Journals Career Network
- Journals on Oxford Academic
- Books on Oxford Academic
Article Contents
Acknowledgements.
- < Previous
DSSPcont: continuous secondary structure assignments for proteins
- Article contents
- Figures & tables
- Supplementary Data
Phil Carter, Claus A. F. Andersen, Burkhard Rost, DSSPcont: continuous secondary structure assignments for proteins, Nucleic Acids Research , Volume 31, Issue 13, 1 July 2003, Pages 3293–3295, https://doi.org/10.1093/nar/gkg626
- Permissions Icon Permissions
The DSSP program automatically assigns the secondary structure for each residue from the three-dimensional co-ordinates of a protein structure to one of eight states. However, discrete assignments are incomplete in that they cannot capture the continuum of thermal fluctuations. Therefore, DSSPcont ( http://cubic.bioc.columbia.edu/services/DSSPcont ) introduces a continuous assignment of secondary structure that replaces ‘static’ by ‘dynamic’ states. Technically, the continuum results from calculating weighted averages over 10 discrete DSSP assignments with different hydrogen bond thresholds. A DSSPcont assignment for a particular residue is a percentage likelihood of eight secondary structure states, derived from a weighted average of the ten DSSP assignments. The continuous assignments have two important features: (i) they reflect the structural variations due to thermal fluctuations as detected by NMR spectroscopy; and (ii) they reproduce the structural variation between many NMR models from one single model. Therefore, functionally important variation can be extracted from a single X-ray structure using the continuous assignment procedure.
Received February 14, 2003; Revised and Accepted April 7, 2003
From discrete to continuous secondary structure assignment.
The automatic assignment of protein secondary structure from three-dimensional co-ordinates of protein structures is an important and, in principle, a simple bioinformatics tool. Assignments are used to visualise structures, to speed up computationally expensive structural comparisons and to improve sequence searches. Secondary structure is more conserved than sequence information. Statistics about secondary structure occurrence can be incorporated into a profile used for homology searches ( 1 , 2 ). This can yield improved accuracy over standard search tools using sequence-based information alone ( 2 – 6 ). Hence, secondary structure assignments are important to assure the optimal yield of experimental structures and to cleverly select the targets for structural genomics. Although a conceptually simple task, the assignment of secondary structure is not always well defined ( 1 ). In fact, assignments vary between different NMR models of the same protein and between X-ray structures of homologues ( 7 ). Previously, we argued that such differences are not a problem of the assignment scheme, rather that they carry important information if adequately processed. Indeed, the variations between different NMR models correlate with thermal disorder ( 7 ). The DSSP program developed by Kabsch and Sander ( 8 ) identifies secondary structure as described by Pauling and colleagues ( 9 , 10 ) for three helix types and two extended sheet types. DSSP has become the standard in the field. DSSPcont constitutes a relatively straightforward extension of DSSP by adding continuous assignments (Fig. 1 ). Because the continuous assignment of secondary structure reproduces the observed variation between high-quality NMR models, it also correlates with mobility related to protein function ( 7 ). Thus, continuous secondary structure assignments can recognise conformational variations from a single X-ray structure and thereby may assist predictions of functionally important residues. More generally, it may help to pave the way to automatically generate valid hypotheses from protein structures. Finally, the continuous assignment appeared to describe ends of regular secondary structure segments (helices and strand) more accurately than discrete assignments. Often these caps carry important information about function and structure. Hence, the continuum may sharpen the tools that already profit from discrete assignments.
Algorithm used to generate DSSPcont.
We assigned a continuum of secondary structure by running DSSP with nine different hydrogen bond thresholds (from −1.0 kcal/mol to −0.2 kcal/mol) ( 7 ). To score a given weighting scheme, we used the different models reported in NMR structure ensembles and calculated the average difference between single model assignments and the mean assignment. The best weighting scheme consequently ensured that the assignment extracted as much information as possible from a single NMR model given. The 100 best weighting schemes were all similar for helix {3 10 -helix, alpha-helix, pi-helix}, strand {extended beta sheet, beta bridge} and other {other/loop, bend, helix-turn}. This similarity indicated that the weighting scheme had a well-defined stable global optimum. The most dominant weights were found close to the default DSSP hydrogen bond threshold of −0.5 kcal/mol. The weight for the −0.2 kcal/mol threshold was consistently low, while the adjacent threshold at −0.3 kcal/mol was consistently high. This prompted us to insert another threshold at −0.25 kcal/mol. To fine-tune the weighting scheme, we performed a simple gradient descent optimisation for 50, 100, 150 and 211 proteins. The DSSPcont assignment is therefore constructed by applying nine hydrogen bond thresholds from −0.2 kcal/mol in steps of 0.1 down to −1 kcal/mol, and in addition the tenth value of −0.25 kcal/mol. The result of the averaging procedure is that a single residue is no longer assigned a single ‘state’, rather the continuous secondary structure of a residue is characterised by a vector with propensities for the eight different DSSP states. More flexible residues have high propensities for more than one ‘state’, while ‘more frozen’ residues have non-zero values only for one particular state. This implies in particular that DSSPcont distinguishes well-defined from rigged helix/strand caps. Furthermore, DSSPcont distinguishes non-regular states that are flexible from those that are not.
Interface to web site.
The DSSPcont server can be accessed through a web interface for use on PDB formatted protein structures ( http://cubic.bioc.columbia.edu/services/DSSPcont ). Users may also access a DSSPcont database of pre-calculated assignments for all PDB ( 11 ) records; this database is updated weekly with all new PDB entries. The interface is very simple, requiring submission of a PDB identifier for the pre-calculated assignments. To run the DSSPcont algorithm on a user's own protein, a file containing the protein can be uploaded or the user can ‘cut and paste’ the protein description into the web interface. The DSSPcont predictions for all PDB entries have been integrated into the database integration system SRS ( 12 ). This enables to search by ‘ID’, ‘Compound Name’, ‘Source’, ‘Author Name’, ‘Number of Residues’, ‘Number of Chains’, ‘Total Number of Disulphide Bridges’, ‘Number of Intrachain Bridges’, ‘Interchain Disulphide Bridges’, ‘Protein Surface Accessibility’, ‘Total Number of Hydrogen Bonds’, ‘Number of Hydrogen Bonds in Parallel Bridges’ and ‘Hydrogen Bonds in Antiparallel Bridges’. The flat files for these DSSPcont assignments can be downloaded and used locally.
Output of DSSPcont.
The algorithm simply adds columns for the continuous assignment (as percentages for each of the eight states distinguished by DSSP) to the DSSP format ( 8 ). DSSP assigns eight states: 3 10 -helix (represented by G), alpha-helix (H), pi-helix (I), helix-turn (T), extended beta sheet (E), beta bridge (B), bend (S) and other/loop (L). Eight columns are added to the standard DSSP output, each representing one of these DSSP states. In the example shown in Figure 1 , there are 23 NMR models for the 1c3y fragment. Figure 1 B shows the DSSPcont assignments for model 1. DSSP assigns the state of other/loop to residue 20 which is a valine. DSSPcont, however, is more detailed, predicting a 68% likelihood that it is involved in other/loop, but also a 32% probability of a helix turn. Each residue in the protein is assigned a percentage for each of the eight states. The core of the helix's residues (24–28) are assigned as H by default DSSP although the entire a-helix switched to a 3 10 -helix when applying a hydrogen bond threshold of −1 kcal/mol. A ‘fuzzy’ helix capping, as seen here, is common and was observed for approximately one in four N-caps and half the C-caps in our data sets. Dissecting the continuous assignment shows that a 0.1 kcal/mol looser hydrogen bond threshold in the default DSSP would extend the helix by one residue (residue 29). If the default threshold instead had been tightened by 0.2 kcal/mol, the helix would lose one residue (residue 28). A more detailed online explanation of the DSSPcont format can be found at http://cubic.bioc.columbia.edu/services/DSSPcont/DSSPcont.html .
Thanks to Chris Sander (Sloan Kettering, New York) for the permission to use DSSP, to Gerrit Vriend (Nijmegen, Netherlands) for maintaining DSSP, to Arthur G. Palmer (Columbia University) and to Søren Brunak (Technical University of Denmark) for their invaluable contributions that were at the base of the scientific development of DSSPcont. Thanks to Jinfeng Liu (Columbia University) for computer assistance. The work was supported by the grants 1-P50-GM62413-01 and RO1-GM63029-01 from the National Institute of Health (NIH). Last, but not least, thanks to all those who deposit their experimental data in public databases and to those who maintain these databases.
![Figure 1. DSSPcont assignment for 1c3y fragment. The variations between the secondary structure assignments for different NMR models of the same protein illustrate the impact of fluctuations on structure and highlight the difficulty of predicting protein structure. (A) The default DSSP assignments for all 23 models of the THP12-carrier protein [PDB identifier 1c3y (8)]. The structure models were calculated using 13C/15N labeled protein and 3D/4D NMR spectroscopy with 13 NOE's per residue. (B) DSSPcont assignments for the first NMR model alone.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/nar/31/13/10.1093_nar_gkg626/2/m_gkg62601.jpeg?Expires=1720713206&Signature=4uLDlsgFv9brcX81Is7d1McVN1E5fvIuAgQShXlrMbDr-k8f47lFJbePA1oCwX8-1~ZMTVUsF08ZTolrfpNBMKS2FmRFZodY3S7DjTA1JCF3yLuDN5cX5Mnbp918006pqhbk~i37gsMXbrd9f3yflQJQxLuYF2Vhz2ywYLKuK9YbwrWWDC5lit6B3iW6Gp7HUm~Nc4V3A1upViTe5yagQ-Kj6xODAm9MphLfATf9F~7KbV9qj1yfFHLMAa22tdwXy6arioU~uthZQxIHixHQepabIbTJGFLCkPfC9h-u6hhNt-yhClptn~ktneJmpBDpJQguVRrqim7jeI6vbv4zQQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA "secondary structure assignment dssp")
Figure 1. DSSPcont assignment for 1c3y fragment. The variations between the secondary structure assignments for different NMR models of the same protein illustrate the impact of fluctuations on structure and highlight the difficulty of predicting protein structure. ( A ) The default DSSP assignments for all 23 models of the THP12-carrier protein [PDB identifier 1c3y ( 8 )]. The structure models were calculated using 13C/15N labeled protein and 3D/4D NMR spectroscopy with 13 NOE's per residue. ( B ) DSSPcont assignments for the first NMR model alone.
Andersen,C.A.F. and Rost,B. ( 2003 ) Secondary structure assignment. In Bourne,P. and Weissig,H. (eds), Structural Bioinformatics. John Wiley, Hoboken, NJ, pp. 339 –361.
Holm,L. and Sander,C. ( 1999 ) Protein folds and families: sequence and structure alignments. Nucleic Acids Res. , 27 , 244 –247.
Jennings,A.J., Edge,C.M. and Sternberg,M.J. ( 2001 ) An approach to improving multiple alignments of protein sequences using predicted secondary structure. Protein Eng. , 14 , 227 –231.
Rost,B. ( 1995 ) TOPITS: threading one-dimensional predictions into three-dimensional structures. In Rawlings,C., Clark,D., Altman,R., Hunter,L., Lengauer,T. and Wodak,S. (eds), Third International Conference on Intelligent Systems for Molecular Biology. Menlo Park, CA: AAAI Press, Cambridge, England, pp. 314 –321.
Rost,B., Schneider,R. and Sander,C. ( 1997 ) Protein fold recognition by prediction-based threading. J. Mol. Biol. , 270 , 471 –480.
Fischer,D. and Eisenberg,D. ( 1996 ) Fold recognition using sequence-derived properties. Protein Sci. , 5 , 947 –955.
Andersen,C.A., Palmer,A.G., Brunak,S. and Rost,B. ( 2002 ) Continuum secondary structure captures protein flexibility. Structure (Camb.) , 10 , 175 –184.
Kabsch,W. and Sander,C. ( 1983 ) Dictionary of protein secondary structure: pattern recognition of hydrogen bonded and geometrical features. Biopolymers , 22 , 2577 –2637.
Pauling,L., Corey,R.B. and Branson,H.R. ( 1951 ) Two hydrogen-bonded helical configurations of the polypeptide chain. Proc. Natl Acad. Sci. USA , 37 , 205 –211.
Pauling,L. and Corey,R.B. ( 1951 ) Configurations of polypeptide chains with favored orientations around single bonds: two new pleated sheets. Proc. Natl Acad. Sci. USA , 37 , 720 –740.
Berman,H.M., Westbrook,J., Feng,Z., Gilliland,G., Bhat,T.N., Weissig,H., Shindyalov,I.N. and Bourne,P.E. ( 2000 ) The Protein Data Bank. Nucleic Acids Res. , 28 , 235 –242.
Etzold,T. and Argos,P. ( 1993 ) SRS—an indexing and retrieval tool for flat file data libraries. Comput. Appl. Biosci. , 9 , 49 –57.
Email alerts
Citing articles via.
- Editorial Board
Affiliations
- Online ISSN 1362-4962
- Print ISSN 0305-1048
- Copyright © 2024 Oxford University Press
- About Oxford Academic
- Publish journals with us
- University press partners
- What we publish
- New features
- Open access
- Institutional account management
- Rights and permissions
- Get help with access
- Accessibility
- Advertising
- Media enquiries
- Oxford University Press
- Oxford Languages
- University of Oxford
Oxford University Press is a department of the University of Oxford. It furthers the University's objective of excellence in research, scholarship, and education by publishing worldwide
- Copyright © 2024 Oxford University Press
- Cookie settings
- Cookie policy
- Privacy policy
- Legal notice
This Feature Is Available To Subscribers Only
Sign In or Create an Account
This PDF is available to Subscribers Only
For full access to this pdf, sign in to an existing account, or purchase an annual subscription.

An official website of the United States government
The .gov means it’s official. Federal government websites often end in .gov or .mil. Before sharing sensitive information, make sure you’re on a federal government site.
The site is secure. The https:// ensures that you are connecting to the official website and that any information you provide is encrypted and transmitted securely.
- Publications
- Account settings
Preview improvements coming to the PMC website in October 2024. Learn More or Try it out now .
- Advanced Search
- Journal List
- Nucleic Acids Res
- v.32(Web Server issue); 2004 Jul 1
STRIDE: a web server for secondary structure assignment from known atomic coordinates of proteins
STRIDE is a software tool for secondary structure assignment from atomic resolution protein structures. It implements a knowledge-based algorithm that makes combined use of hydrogen bond energy and statistically derived backbone torsional angle information and is optimized to return resulting assignments in maximal agreement with crystallographers' designations. The STRIDE web server provides access to this tool and allows visualization of the secondary structure, as well as contact and Ramachandran maps for any file uploaded by the user with atomic coordinates in the Protein Data Bank (PDB) format. A searchable database of STRIDE assignments for the latest PDB release is also provided. The STRIDE server is accessible from http://webclu.bio.wzw.tum.de/stride/ .
INTRODUCTION
Identification of secondary structure elements is a major step in the characterization of a newly determined protein structure. It serves as a basis for virtually all subsequent analyses, including visualization, structure comparison and classification, homology modelling, threading and sequence alignment. To a large extent, our visual notion of proteins is based on cartoon diagrams showing α-helices and β-strands as cylinders and arrows, respectively.
Several automatic tools for secondary structure assignment from known atomic coordinates are available [reviewed in ( 1 )]. The most widely used method, DSSP ( 2 ), defines secondary structure elements as repeating elementary hydrogen bonded patterns. Hydrogen bonds between peptide units are assigned if the electrostatic interaction energy between C=O of one residue and N–H of another residue is <−0.5 kcal/mole. The DEFINE algorithm ( 3 ) compares inter-atomic distance matrices of structural fragments to idealized reference distance masks typical for a particular secondary structure type, while P-Curve ( 4 ) is based on quantification of backbone curvature using differential geometry. More recently, an improved version of DSSP, called DSSPcont, has been developed which takes into account the structural variations in proteins ( 5 ).
Our method, STRIDE ( 6 ), was developed with a specific goal to accurately reproduce secondary structure designations created by human experts. It is thus a knowledge-based approach which uses, as training data, a carefully verified set of secondary structural elements defined by crystallographers who have deposited structures in the Protein Data Bank (PDB) ( 7 ). The main difference between STRIDE and DSSP is that STRIDE considers both hydrogen bonding patterns and backbone geometry. The hydrogen bond energy is calculated using an empirical energy function which takes into account the distance between the donor and the acceptor and the deviations from linearity of the bond angles ( 8 , 9 ). A weighted product of hydrogen bond energy and torsion angle probabilities for α-helix and β-sheet is used to determine the start and stop positions of secondary structure elements based on empirically optimized recognition thresholds.
The source code of STRIDE has been freely accessible from the FTP server of the European Bioinformatics Institute since 1995 ( ftp://ftp.ebi.ac.uk/pub/software/unix/stride/src ). It is also available as part of several molecular graphics packages and websites [e.g. Visual Molecular Dynamics (VMD) ( 10 )]. Here, we report a dedicated STRIDE web server and a database of secondary structure assignments.
STRIDE WEB SERVER AND DATABASE
The STRIDE web server, written in the python programming language, makes accessible all functions implemented in the STRIDE software and also provides several additional visualization tools (Figure (Figure1). 1 ). It accepts as input atomic coordinates in standard PDB format, which can be either uploaded or pasted into a web form. The STRIDE home page offers the following options.

Available views of the secondary structure assignment created by STRIDE from the sample structure 456c. Center top, original STRIDE output; left, contact map; right, Ramachandran plot; bottom, cartoon representation of secondary structure.
Basic secondary structure assignment . A secondary structure assignment in text form is produced. Its header section gives general information about the structure (author, compound, etc.) as well as a secondary structure summary which lists locations of identified secondary structure elements. Then follows a detailed per residue assignment of secondary structure states complemented by information on backbone dihedral angles and solvent accessible area computed according to Eisenhaber et al . ( 11 , 12 ).
Graphical representation of secondary structure assignment . A cartoon representation similar to the ‘wiring diagram’ of the PDBsum web server ( 13 ) is produced. Technically, this representation is generated not as a rendered image or postscript file, but rather in the form of an html table incorporating both individual graphical items and the protein sequence. The table is constructed by parsing the output of STRIDE and assigning an image to each structural state. An interactive point-and-click interface reveals detailed per residue information.
Contact map . A contact map is derived from a symmetric square matrix of distances between all C-α atoms in a given protein. An interactive mouse-sensitive image indicates distances below a certain threshold defined by the user, typically 6 Å.
Ramachandran plot . No secondary structure web server is complete without a Ramachandran plot ( 14 ) representing the distribution of ϕ and φ torsion angles in a given protein. The allowed areas for α-helix and β-sheet are shown in the background of the plot. They are taken from a recent update of the Ramachandran map by Lovell et al . ( 15 ). The orange line is the border for the core region of favourable angles. Gold limits the region of disfavoured but allowed angles. Like the contact map, this image is also mouse sensitive and gives additional information on the residue that is pointed at.
Database of STRIDE assignments . A complete database of STRIDE secondary structure assignments is calculated from each weekly update of PDB ( 7 ). Individual entries can be accessed either by PDB code or through a text search interface allowing for the construction of logically structured queries.
pydssp 0.9.0
pip install pydssp Copy PIP instructions
Released: Nov 15, 2022
A simplified implementation of DSSP algorithm for PyTorch and NumPy
Verified details
Maintainers.

Unverified details
Project links, github statistics.
- Open issues:
View statistics for this project via Libraries.io , or by using our public dataset on Google BigQuery
License: MIT License (MIT)
Author: Shintaro Minami
Tags DSSP, Secondary Structure, Protein Structure
Classifiers
- OSI Approved :: MIT License
- Python :: 3.8
Project description
What's this.
DSSP (Dictionary of Secondary Structure of Protein) is a popular algorithm for assigning secondary structure of protein backbone structure. [ Wolfgang Kabsch, and Christian Sander (1983) ] This repository is a python implementation of DSSP algorithm that simplifies some parts of the algorithm.
General Info
- It's NOT a complete implementation of the original DSSP, as some parts have been simplified (some more details here ). However, an average of over 97% of secondary structure determinations agree with the original.
- The algorithm used to identify hydrogen bonded residue pairs is exactly the same as the original DSSP algorithm, but is extended to output the hydrogen-bond-pair-matrix as continuous values in the range [0,1].
- With the continuous variable extension above, the hydrogen-bond-pair-matrix is differentiable with torch.Tensor as input.
install through PyPi
Install by git clone, to use pydssp script.
If you have already installed pydssp, you should be able to use pydssp command.
The output.result will be a text format, looking like follows,
To use as python module
Import & test coordinates, to get hydrogen-bond matrix: pydssp.get_hbond_map().
- For hbond_matrix[b, i, j], index 'i' is for donner (N-H) and 'j' is for acceptor (C=O), respectively
- The output matrix consists of constant values in the range [0,1], which is defined as follows.
$HbondMat(i,j) = (1+\sin((-0.5-E(i,j)-margin)/margin*\pi/2))/2$
Here $E$ is the electrostatic energy defined by (Kabsch and Sander 1983) and $margin(=1.0)$ is introduced to control smoothness.
To get secondary structure assignment: pydssp.assign()
Differences from the original dssp.
This implementation was simplified from the original DSSP algorithm. The differences from the original DSSP are as follows
- The implementation omitted β-bulge annotation, so β-bulge is determined as a loop instead of β-strand.
- Parameters for adding hydrogen atoms are slightly different from the original DSSP, which may cause small differences in hydrogen bond annotation.
- Only support C3 ('-', 'H', and 'E') type assignment instead of C8 type (B, E, G, H, I, S, T, and ' ').
Although the above simplifications, the C3 type annotation still matches with the original DSSP for more than 97% of residues on average.
Project details
Release history release notifications | rss feed.
Nov 15, 2022
Nov 14, 2022
Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages .
Source Distributions
Built distribution.
Uploaded Nov 15, 2022 Python 3
Hashes for pydssp-0.9.0-py3-none-any.whl
- português (Brasil)
Supported by


Calculate secondary structure content using the DSSP algorithm
[<name>] Output data set name. [out <filename>] Output file name for secondary structure vs time. [<mask>] Atom mask in which residues should be looked for. [sumout <sumfilename>] Write average secondary structure values for each residue to <sumfilename>; if not specified <filename>.sum is used. [assignout <filename>] Write overall secondary structure assignment (based on dominant secondary structure type for each residue) to file. [ptrajformat] Write secondary structure as a string of characters for each frame, similar to ptraj output. [betadetail] Record anti-parallel beta and parallel beta in place of extended and bridge secondary structure. If a residue could be both only anti-parallel is reported. [namen <N name>] Backbone amide nitrogen atom name (default ’N’). [nameh <H name>] Backbone amide hydrogen atom name (default ’H’). [nameca <CA name>] Backbone alpha carbon atom name (default ’CA’). [namec <C name>] Backbone carbonyl carbon atom name (default ’C’). [nameo <O name>] Backbone carbonyl oxygen atom name (default ’O’). [namesg <SG name>] Cysteine sulfur atom name, used to ignore disulfide connectivity (default ‘SG’).
Data Sets Created: <name>[res] Residue secondary structure per frame; index corresponds to residue number. If ptrajformat specified these will be characters, otherwise integers (see table below). <name>[avgss] Average of each type of secondary structure; index corresponds to secondary structure type (see table below; no index for “None”). <name>[None] Total fraction of residues with no structure vs time. <name>[Para] Total fraction of residues with parallel beta structure vs time. <name>[Anti] Total fraction of residues with anti-parallel beta structure vs time. <name>[3-10] Total fraction of 3-10 helical structure vs time. <name>[Alpha] Total fraction of alpha helical structure vs time. <name>[Pi] Total fraction of Pi helical structure vs time. <name>[Turn] Total fraction of turn structure vs time. <name>[Bend] Total fraction of bend structure vs time.
As of version 4.18.0, this command now produces output that better conforms with the original deinitions in Kabsch and Sander ; namely that Extended beta (i.e. 2 or more consecutive beta bridges of the same type) and beta Bridge (i.e. an isolated beta bridge) are now reported instead of anti-parallel and parallel beta. To restore the original behavior the betadetail keyword must be specified
Note that when not using ptraj format, data sets are not generated until run is called.
Calculate secondary structural propensities for residues in <mask> (or all solute residues if no mask given) using the DSSP method of Kabsch and Sander , which assigns secondary structure types for residues based on backbone amide (N-H) and carbonyl (C=O) atom positions. By default CPPTRAJ assumes these atoms are named “N”, “H”, “C”, and “O” respectively. If a different naming scheme is used (e.g. amide hydrogens are named “HN”) the backbone atom names can be customized with the nameX keywords (e.g. ’nameH HN’). Note that it is expected that some residues will not have all of these atoms (such as proline); in this case CPPTRAJ will print an informational message but the calculation will proceed normally.
Results will be written to filename specified by out with format:
<#Frame> <ResX SS> <ResX+1 SS> ... <ResN SS>
where <#Frame> is the frame number and <ResX SS> is an integer representing the calculated secondary structure type for residue X. If the keyword ptrajformat is specified, the output format will instead be:
<#Frame> STRING
where STRING is a string of characters (one for each residue) where each character represents a different structural type (this format is similar to what ptraj outputs). The various secondary structure types and their corresponding integer/character are listed below:
Average structural propensities over all frames for each residue will be written to the file specified by sumout (or “<filename>.sum” if sumout is not specified). The total structural propensity over all residues for each secondary structure type will be written to the file specified by totalout . If assignout is specified, the overall secondary structure assignment for each residue will be printed in two line chunks of 50 residues, with the first line containing the residue number the line starts with and one character residue names, and the second line containing secondary structure assignment using DSSP-style characters, like so:
The output of secstruct command is amenable to visualization with gnuplot. To generate a 2D map-style plot of secondary structure vs time, with each residue on the Y axis simply give the output file a “.gnu” extension.
For example, to generate a 2D map of secondary structure vs time, with different colors representing different secondary structure types for residues 1-22:
The resulting file can be visualized with gnuplot: gnuplot dssp.gnu Similarly, the sumout file can be nicely visualized using xmgrace (use “.agr” extension).
Values of dgbulk and dhbulk for different water models can be calculated from pure water simulations with the purewater keyword.
Tags: action commands , analysis
Copyright © 2024 · All Rights Reserved · AMBER-hub
Theme: Structure Lite by Organic Themes
IMAGES
VIDEO
COMMENTS
Introduction. The DSSP program was designed by Wolfgang Kabsch and Chris Sander to standardize secondary structure assignment. DSSP is a database of secondary structure assignments (and much more) for all protein entries in the Protein Data Bank (PDB). DSSP is also the program that calculates DSSP entries from PDB entries.
The DSSP algorithm is the standard method for assigning secondary structure to the amino acids of a protein, given the atomic-resolution coordinates of the protein. The abbreviation is only mentioned once in the 1983 paper describing this algorithm, where it is the name of the Pascal program that implements the algorithm Define Secondary Structure of Proteins.
The DSSP program was designed by Wolfgang Kabsch and Chris Sander to standardize secondary structure assignment. DSSP is a database of secondary structure assignments (and much more) for all protein entries in the Protein Data Bank (PDB). DSSP is also the program that calculates DSSP entries from PDB entries.
The online DSSP program calculates DSSP (Define Secondary Structure of Proteins) entries from PDB entries. DSSP is the standard method for assigning secondary structure to the amino acids of a protein, given the atomic coordinates of the protein. Eight types of secondary structure are assigned in this program.
Several automatic tools for secondary structure assignment from known atomic coordinates are available [reviewed in ]. The most widely used method, DSSP , defines secondary structure elements as repeating elementary hydrogen bonded patterns. Hydrogen bonds between peptide units are assigned if the electrostatic interaction energy between C=O of ...
A few years later, Kabsch and Sander developed a method called DSSP that still remains one of the most widely-used program for secondary structure assignment. The DSSP algorithm is based on the detection of hydrogen-bonds defined by an electrostatic criterion.
A first step toward three-dimensional protein structure description is the characterization of secondary structure. The most widely used program for secondary structure assignment remains DSSP, introduced in 1983, with currently more than 400 citations per year. DSSP output is in a one-letter repres …
DSSP provides an elaborate description of the secondary structure elements in a protein structure, including backbone hydrogen bonding and the topology of β-sheets. The most popular feature is the per-residue assignment of secondary structure with a single character code: H = α-helix. B = residue in isolated β-bridge.
A DSSPcont assignment for a particular residue is a percentage likelihood of eight secondary structure states, derived from a weighted average of the ten DSSP assignments. The continuous assignments have two important features: (i) they reflect the structural variations due to thermal fluctuations as detected by NMR spectroscopy; and (ii) they ...
DSSP (Dictionary of Protein Secondary Structures) and STRIDE (STructural IDEntification) are two gold standards in protein structure assignment. DSSP works on fine-grained protein structures and confirms a hydrogen bond when the electrostatic energy (E) between each interacting pair is less than −0.5 kcal/mole [ 9 ].
The variations between the secondary structure assignments for different NMR models of the same protein illustrate the impact of fluctuations on structure and highlight the difficulty of predicting protein structure. (A) The default DSSP assignments for all 23 models of the THP12-carrier protein [PDB identifier 1c3y ]. The structure models were ...
The secondary structure is assigned based on hydrogen bonding patterns as those initially proposed by Pauling et al. in 1951 (before any protein structure had ever been experimentally determined). There are eight types of secondary structure that DSSP defines: G = 3-turn helix ( 3 10 helix ). Min length 3 residues.
In PDB format, secondary structure assignments are described in HELIX and SHEET records, and analogous information is included in mmCIF files. However, not all input files include this information. The dssp calculation is run: when an atomic structure of a protein without secondary structure assignments is read
dssp - Secondary structure assignment¶ Introduction¶. DSSP is a program developed by Wolfgang Kabsch and Chris Sander to assign secondary structure states to protein structures. The assignment is based on hydrogen bonding patterns and geometric features.
Several automatic tools for secondary structure assignment from known atomic coordinates are available [reviewed in ]. The most widely used method, DSSP , defines secondary structure elements as repeating elementary hydrogen bonded patterns. Hydrogen bonds between peptide units are assigned if the electrostatic interaction energy between C=O of ...
DSSPcont is a variant of DSSP aiming to assign secondary structure in a continuous fashion, thus reflecting the structural variability due to thermal motion. ... The net result is over-assignment of secondary structure compared to other methods with approximately 80% of residues being assigned a regular secondary structure class. While over ...
Of these, DSSP and STRIDE use quite close criteria for the assignment of secondary structure and are the most widely used methods. The DSSP (Dictionary of Secondary Structure of Proteins) method, developed by Kabsch and Sander, uses hydrogen bond patterns based on an electrostatic model to assign the secondary structures.
DSSP (Dictionary of Secondary Structure of Protein) is a popular algorithm for assigning secondary structure of protein backbone structure. [Wolfgang Kabsch, and ... type assignment instead of C8 type (B, E, G, H, I, S, T, and ' '). Although the above simplifications, the C3 type annotation still matches with the original DSSP for more than 97% ...
If assignout is specified, the overall secondary structure assignment for each residue will be printed in two line chunks of 50 residues, with the first line containing the residue number the line starts with and one character residue names, and the second line containing secondary structure assignment using DSSP-style characters, like so: